Általánosság
A retinoblasztóma (Rb) rosszindulatú szemdaganat, amely a retina sejtjeiből fejlődik ki. Ez a rák bármely életkorban előfordulhat, de a leggyakoribb az ötéves kor előtti csecsemőkorban.

A gyermekkori rák agresszív: a retinoblasztóma átterjedhet a nyirokcsomókra, a csontokra vagy a csontvelőre. Ritkán a központi idegrendszert (agy és gerincvelő) érinti.
A retinoblasztómában szenvedő gyermekek körülbelül 90% -ának pozitív a prognózisa (a gyógyulás valószínűsége), feltéve, hogy a diagnózis korai, és a kezelést még a rák terjedése előtt megkezdik. Amikor csak lehetséges, az orvosi beavatkozás célja a beteg látásának megőrzése.
Okoz
A daganat kialakulásához vezető események sora összetett: ez akkor kezdődik, amikor a retina sejtjeiben mutáció (vagy deléció) alakul ki, amely magában foglalja az RB1 tumorszuppresszor gént, amely a 13. kromoszóma q14 sávján (13q14) található.
Minden sejt általában két RB1 gént tartalmaz:
- Ha a gén legalább egy példánya megfelelően működik, a retinoblasztóma nem keletkezik (de a kockázat nő);
- Ha a gén mindkét példánya mutálódik vagy hiányzik, ellenőrizetlen sejtproliferáció következik be.
Sok esetben nem világos, hogy pontosan mi váltja ki az RB1 gén változásait (szórványos retinoblasztóma); ezek véletlen genetikai hibákból eredhetnek, amelyek például a szaporodás és a sejtosztódás során jelentkeznek. Ismeretes azonban, hogy a retinoblasztóma mögött meghúzódó genetikai rendellenességek szülőktől gyermekekre is továbbadhatók, autoszomális domináns öröklődési mintával. Ez azt jelenti, hogy ha egy szülő mutált (domináns) gént hordoz, akkor minden gyermek 50% -os esélyt kap arra, hogy örökölje, és 50% -kal esélye lesz normális genetikai felépítésre (recesszív gének).
- Egy alkalmi sejt inaktiválja az egyetlen normális másolatát az RB1 génből (egy példány már mutálódott);
- Az RB1 két példányának elvesztése a retina túlzott proliferációjához vezet.
- Egy alkalmi sejt inaktiválja egyik normális RB1 génjét;
- Az RB1 gén második példánya inaktivált;
- Az RB1 két példányának elvesztése túlzott sejtproliferációt indukál, ami retinoblasztómához vezet.
Genetikai és molekuláris jellemzők
- A retinoblasztóma volt az első daganat, amely közvetlenül kapcsolódott "genetikai rendellenességhez (a 13. kromoszóma q14 sávjának törlése vagy mutációja).
- Az RB1 kódolja a pRb fehérjét, amely kulcsszerepet játszik a sejtciklusban: lehetővé teszi a DNS replikációját és a sejtciklus előrehaladását, mivel részt vesz az S fázisú gének (G1 → † "S) transzkripciós szabályozásában.
- A retinoblasztóma mellett az RB1 gén inaktiválódik a hólyag-, emlő- és tüdőrákban.
Örökletes retinoblasztóma
Az örökletes retinoblasztómában szenvedő gyermekeknél a betegség hajlamosabb a korai életkorra, mint a szórványos esetek. Ezenkívül ezeknek a gyermekeknek nagyobb a kockázata más nem szemészeti rákos megbetegedéseknek, mivel az RB1 gén rendellenessége veleszületett (azaz születéstől fogva jelen van), és a test összes sejtjét érinti (csíravonal-mutáció néven ismert), beleértve mindkettőt. retina: Emiatt az örökletes formájú gyermekek gyakran kétoldali retinoblasztómában szenvednek, nem csak egy szemben.
Tünetek
További információ: Retinoblastoma tünetei
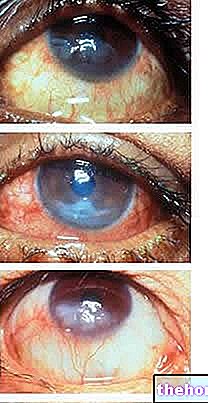
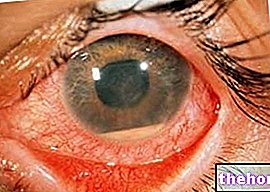
A retinoblasztóma leggyakoribb és legnyilvánvalóbb jele a pupilla rendellenes megjelenése, amely szürkésfehér visszaverődést mutat, amikor fénysugár éri (leukokória vagy amaurotikus macskareflex)). Egyéb jelek és tünetek: csökkent látás, szemfájdalom és -vörösség, valamint fejlődési késleltetés. Néhány retinoblasztómában szenvedő gyermeknél hunyorodás (rosszul beállított szemek) alakulhat ki; más esetekben lehetséges a neovaszkuláris glaukóma megtalálása, amely egy idő után a szem megnagyobbodását (buftalmo) okozhatja.
A rákos sejtek tovább hatolhatnak a szembe és más struktúrákba:
- Intraokuláris retinoblasztóma. A retinoblasztóma intraokulárisan definiálható, ha a daganat teljes egészében a szem belsejében helyezkedik el. A daganat csak a retinában található, vagy más részeket is érint, például a koroidát, a csillóstestet és a látóideg egy részét. Ezért az intraokuláris retinoblasztóma nem terjed a szem külső szöveteire.
- Extraocularis retinoblastoma.A daganat elszaporodhat, és befolyásolhatja a szem körüli szöveteket (orbitális retinoblasztóma). A rák a test más területeire is átterjedhet, például az agyra, a gerincre, a csontvelőre és a nyirokcsomókra (metasztatikus retinoblasztóma).
Az orbitális kiterjedés, az uveális érintettség és a látóideg inváziója ismert kockázati tényező a metasztatikus retinoblasztóma kialakulásához.
Diagnózis
Pozitív családi anamnézis esetén a páciens rendszeres szemvizsgálaton megy keresztül a rák szűrésére. Ha a veleszületett retinoblasztóma kétoldalú, akkor általában az első életévben diagnosztizálják, míg ha csak az egyik szemet érinti, akkor a daganat jelenléte körülbelül 18-30 hónapos korban igazolható.
A retinoblasztóma klinikai diagnózisát a szemfenék vizsgálata határozza meg.A daganat a helyétől függően a szem egyszerű vizsgálata során látható lehet, közvetett oftalmoszkópiával. Képalkotó technikák alkalmazhatók a diagnózis megerősítésére, a daganat stádiumának meghatározására (hol van, mennyire elterjedt, befolyásolja -e a test más szerveinek működését stb.), És megállapítja, hogy a kezelés eredményes volt -e . A vizsgálatok közé tartozhat az ultrahang, a számítógépes tomográfia (CT) és a mágneses rezonancia képalkotás (MRI).
A molekuláris-genetikai diagnózis az RB1 gén mutációjának azonosításával lehetséges. A perifériás vér limfocitáinak citogenetikai analízisét (azaz a kromoszómák) a 13. kromoszóma (13q14.1-q14. 2) delécióinak vagy átrendeződéseinek kimutatására használják. .
Kezelések
Retinoblasztóma esetén számos kezelési lehetőség alkalmazható.
A kezelés céljai a következők:
- Távolítsa el a daganatot és mentse a beteg életét;
- Ha lehetséges, mentse a szemet;
- A látást a lehető legnagyobb mértékben megőrizni;
- Kerülje más rákos megbetegedések kialakulását, amelyeket szintén okozhat a kezelés, különösen örökletes retinoblasztómában szenvedő gyermekeknél.
A prognózis (a gyógyulás valószínűsége) és a kezelési lehetőségek a következő tényezőktől függenek:
- A daganat stádiuma;
- A beteg kora és általános egészségi állapota;
- A daganat gócok elhelyezkedése, mérete és száma;
- A rák terjedése a szemgolyón kívül más területekre is
- Mennyire valószínű, hogy a látás megmaradhat egyik vagy mindkét szemében.
A legtöbb retinoblasztóma esetet korán diagnosztizálják és sikeresen kezelik, mielőtt a rák a szemgolyón kívül áttétet okozhat, ami 90%-os gyógyulási arányt eredményez.



.jpg)













.jpg)


