Hatóanyagok: Erlotinib
Tarceva 25 mg filmtabletta
Tarceva 100 mg filmtabletta
Tarceva 150 mg filmtabletta
Miért alkalmazzák a Tarceva -t? Mire való?
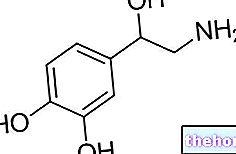
A Tarceva hatóanyaga az erlotinib. A Tarceva egy rák kezelésére használt gyógyszer, amely gátolja az epidermális növekedési faktor receptor (EGFR) nevű fehérje aktivitását, amely részt vesz a rákos sejtek növekedésében és terjedésében.
A Tarceva felnőtteknek javallt. Ezt a gyógyszert felírhatják Önnek, ha előrehaladott nem kissejtes tüdőrákban szenved. Felírható Önnek kezdeti terápiaként vagy terápiaként, ha a betegség a kezdeti kemoterápia után többnyire változatlan marad, mindaddig, amíg a rákos sejtek specifikus EGFR -mutációkkal rendelkeznek. Akkor is felírható, ha a korábbi kemoterápia nem tudta megállítani a rákot . betegség.
Ezt a gyógyszert más gemcitabin nevű kezeléssel kombinálva is felírhatják Önnek, ha áttétes hasnyálmirigyrákja van.
Ellenjavallatok Amikor a Tarceva -t nem szabad alkalmazni
Ne szedje a Tarceva -t
- ha allergiás az erlotinibre vagy a gyógyszer (6. pontban felsorolt) egyéb összetevőjére.
Az alkalmazással kapcsolatos óvintézkedések Mit kell tudnia a Tarceva szedése előtt
Figyelmeztetések és óvintézkedések:
- ha más gyógyszereket szed, amelyek növelhetik vagy csökkenthetik az erlotinib mennyiségét a vérben, vagy befolyásolhatják annak hatékonyságát (például gombaellenes szerek, például ketokonazol, proteázgátlók, eritromicin, klaritromicin, fenitoin, karbamazepin, barbiturátok, rifampicin, ciprofloxacin, omeprazol, ranitidin, orbáncfű vagy proteaszóma inhibitorok), kérdezze meg kezelőorvosát. Bizonyos esetekben ezek a gyógyszerek csökkenthetik a Tarceva hatékonyságát vagy fokozhatják a mellékhatásokat, és ha igen, akkor orvosának módosítania kell a kezelést. Orvosa elrendelheti, hogy ne szedje ezeket a gyógyszereket a Tarceva -kezelés alatt.
- ha véralvadásgátlókat (trombózist vagy véralvadást megelőző gyógyszereket, például warfarint) szed, a Tarceva fokozhatja a vérzésre való hajlamot.Beszéljen kezelőorvosával, akinek rendszeres vérvizsgálatok felírásával ellenőriznie kell Önt.
- ha sztatinokat (a vér koleszterinszintjét csökkentő gyógyszereket) szed, a Tarceva növelheti a sztatinokkal kapcsolatos izomproblémák kockázatát, ami ritka esetekben súlyos izomleépüléshez (rabdomiolízis) vezethet, ami vesekárosodáshoz vezethet. Beszéljen orvosával.
- ha kontaktlencsét használ, és / vagy valaha volt olyan szemproblémája, mint a súlyos szemszárazság, a szem elülső részének (szaruhártya) gyulladása vagy a szem elülső részét érintő fekélyek, forduljon orvosához. Lásd még "Egyéb gyógyszerek és a Tarceva".
Tájékoztatnia kell orvosát:
- ha „hirtelen fellépő légzési nehézségei vannak köhögéssel vagy lázas betegséggel összefüggésben, mivel kezelőorvosának esetleg más gyógyszereket kell felírnia, és abba kell hagynia a Tarceva szedését;
- ha hasmenése van, mivel orvosának esetleg hasmenés elleni gyógyszereket kell felírnia (pl. loperamid);
- azonnal, ha súlyos vagy tartós hasmenése, hányingere, étvágytalansága vagy hányása van, mivel előfordulhat, hogy orvosának abba kell hagynia a Tarceva -kezelést, és kórházi kezelésre van szüksége.
- - ha súlyos hasi fájdalmai, súlyos bőrreakciói vannak, például hólyagosodás vagy hámlás.
- ha akut vörösség vagy súlyosbodó szempír jelentkezik, amelyet fájdalom, fokozott könnyezés, homályos látás és / vagy fényérzékenység kísér, azonnal forduljon orvosához vagy a nővérhez, mert sürgős kezelésre lehet szüksége (lásd Lehetséges mellékhatások).
- ha sztatint is szed, és megmagyarázhatatlan izomfájdalmat, érzékenységet, gyengeséget vagy görcsöket tapasztal. Az orvos szükségesnek találhatja a kezelés megszakítását vagy felfüggesztését.
Lásd még a 4. pontot „Lehetséges mellékhatások”.
A máj és a vesék betegségei
Nem ismert, hogy a Tarceva hatása megváltozik -e, ha a máj vagy a vesék nem működnek megfelelően. Ha Ön súlyos máj- vagy vesebetegségben szenved, nem ajánlott ezzel a gyógyszerrel kezelni.
Glükuronációs rendellenesség, például Gilbert -szindróma
Ha glükuronidációs rendellenessége van, például Gilbert -szindróma, kezelőorvosának óvatosan kell kezelnie.
Füst
Ha Tarceva -t szed, hagyja abba a dohányzást, mivel a dohányzás csökkentheti a gyógyszer mennyiségét a vérében.
Gyermekek és serdülők
A Tarceva -t nem vizsgálták 18 év alatti betegeknél. A gyógyszer alkalmazása gyermekeknél és serdülőknél nem ajánlott.
Kölcsönhatások Mely gyógyszerek vagy élelmiszerek módosíthatják a Tarceva hatását
Egyéb gyógyszerek és a Tarceva
Feltétlenül tájékoztassa kezelőorvosát vagy gyógyszerészét a jelenleg vagy nemrégiben szedett, valamint szedni tervezett egyéb gyógyszereiről.
A Tarceva egyidejű bevétele étellel és itallal
Ne vegye be a Tarceva -t étellel. Lásd még a 3. pontot „Hogyan kell szedni a Tarceva -t”?
Figyelmeztetések Fontos tudni, hogy:
Terhesség és szoptatás
Kerülje a teherbeesést a Tarceva -kezelés alatt. Ha úgy gondolja, hogy terhes lehet, alkalmazzon megfelelő fogamzásgátlást a kezelés alatt és legalább 2 hétig az utolsó tabletta bevétele után. Ha terhes, úgy gondolja, hogy terhes, vagy terhességet tervez, vagy ha szoptat, kérje ki kezelőorvosa vagy gyógyszerésze tanácsát, mielőtt elkezdi szedni ezt a gyógyszert.
A készítmény hatásai a gépjárművezetéshez és gépek kezeléséhez szükséges képességekre
Nem végeztek vizsgálatokat a Tarceva gépjárművezetéshez és gépek kezeléséhez szükséges képességekre gyakorolt lehetséges hatásaival kapcsolatban, de a kezelés nem valószínű, hogy megváltoztatja ezt a képességet.
Túlérzékenység
A Tarceva laktóz -monohidrát nevű cukrot tartalmaz. Ha kezelőorvosa korábban már figyelmeztette Önt, hogy bizonyos cukrokra érzékeny, keresse fel orvosát, mielőtt elkezdi szedni a Tarceva -t.
Adagolás, az alkalmazás módja és ideje A Tarceva alkalmazása: Adagolás
Ezt a gyógyszert mindig az orvos által elmondottaknak megfelelően szedje. Ha kétségei vannak, forduljon orvosához vagy gyógyszerészéhez.
A tablettát legalább egy órával étkezés előtt vagy két órával étkezés után kell bevenni.
A szokásos adag naponta egy Tarceva 150 mg tabletta, ha nem kissejtes tüdőrákja van. A metasztázisos hasnyálmirigyrák esetén a szokásos adag napi egy 100 mg -os Tarceva tabletta. A Tarceva -t gemcitabinnal kombinációban alkalmazzák.
Kezelőorvosa egyszerre 50 mg -mal módosíthatja az adagot. A különböző adagolási rendekhez a Tarceva 25 mg, 100 mg vagy 150 mg erősségben kapható.
Túladagolás Mi a teendő, ha túl sok Tarceva -t vett be?
Ha az előírtnál több Tarceva -t vett be
Azonnal forduljon orvosához vagy gyógyszerészéhez. Előfordulhat, hogy a mellékhatások rosszabbodnak, és kezelőorvosa le fogja állítani a szedését.
Ha elfelejtette bevenni a Tarceva -t
Ha kihagy egy vagy több Tarceva adagot, a lehető leghamarabb forduljon orvosához vagy gyógyszerészéhez. Ne vegyen be kétszeres adagot a kihagyott adag pótlására.
Ha idő előtt abbahagyja a Tarceva szedését
Fontos, hogy minden nap folytassa a Tarceva szedését mindaddig, amíg orvosa előírja.
Ha bármilyen további kérdése van a gyógyszer alkalmazásával kapcsolatban, kérdezze meg kezelőorvosát vagy gyógyszerészét.
Mellékhatások Melyek a Tarceva mellékhatásai?
Mint minden gyógyszer, így ez a gyógyszer is okozhat mellékhatásokat, amelyek azonban nem mindenkinél jelentkeznek.
Ha az alább felsorolt mellékhatások bármelyikét észleli, forduljon orvosához a lehető leghamarabb. Bizonyos esetekben az orvosnak csökkentenie kell a Tarceva adagját vagy le kell állítania a kezelést.
- Hasmenés és hányás (nagyon gyakori, 10 beteg közül több mint 1 beteget érinthet). A tartós és súlyos hasmenés csökkent vérkáliumhoz és veseelégtelenséghez vezethet, különösen akkor, ha más kemoterápiás gyógyszerekkel egyidejűleg kezelik. Súlyosabb vagy tartós hasmenés esetén azonnal forduljon orvosához, aki úgy dönthet, hogy kórházi kezelésben részesül.
- A szemirritáció a kötőhártya -gyulladás / keratoconjunctivitis (nagyon gyakori, 10 beteg közül több mint 1 beteget érinthet) és a keratitis (gyakori, 10 beteg közül legfeljebb 1 -et érinthet) miatt.
- A tüdőgyulladás egyik formája, az úgynevezett intersticiális tüdőbetegség (európai betegeknél nem gyakori, japán betegeknél gyakori: 100 -ból legfeljebb 1 beteget érinthet Európában, és legfeljebb 10 -ből Japánban). Ez a betegség az egészségi állapotának természetes előrehaladásához is köthető, és bizonyos esetekben halálos is lehet. Ha olyan tüneteket tapasztal, mint "hirtelen fellépő légzési nehézség, amely köhögéssel vagy lázhoz kapcsolódik, azonnal forduljon orvosához, mert előfordulhat, hogy ez a betegsége van. Orvosa dönthet úgy, hogy véglegesen leállítja a Tarceva -kezelést."
- Emésztőrendszeri perforációt észleltek (nem gyakori, 100 beteg közül legfeljebb 1 beteget érinthet). Tájékoztassa kezelőorvosát, ha erős fájdalmat érez a hasában, továbbá tájékoztassa kezelőorvosát, ha valaha is volt peptikus fekélye vagy divertikuláris betegsége, mivel ez növelheti a perforáció kockázatát.
- Ritka esetekben májelégtelenséget figyeltek meg (ritka, 1000 betegből legfeljebb 1 beteget érinthet).
Nagyon gyakori mellékhatások (10 beteg közül több mint 1 beteget érinthet):
- Bőrkiütés, amely kialakulhat vagy súlyosbodhat a napsugárzásnak kitett területeken. Ha napfénynek van kitéve, tanácsos védőruházatot és / vagy fényvédőt használni (pl. Ásványi anyagok alapján)
- Fertőzés
- Étvágytalanság, fogyás
- Depresszió
- Fejfájás, a bőrérzet megváltozása vagy a végtagok zsibbadása
- Légzési nehézség, köhögés
- Hányinger
- Szájirritációk
- Gyomorfájdalom, emésztési zavarok és puffadás
- Változások a májfunkcióval kapcsolatos vérvizsgálatokban
- Viszkető, száraz bőr és hajhullás
- Fáradtság, láz, hidegrázás
Gyakori mellékhatások (10 beteg közül legfeljebb 1 beteget érinthet):
- Orrvérzés
- Vérzés a gyomorból vagy a belekből
- Gyulladásos reakciók a körmök körül
- Szőrtüsző fertőzés
- Pattanás
- Hasított bőr (bőrrepedések)
- Csökkent vesefunkció (ha a jóváhagyott javallatokon kívül adják kemoterápiával kombinálva)
Nem gyakori mellékhatások (100 beteg közül legfeljebb 1 beteget érinthet)
- a szempillák elváltozásai
- túlzott szőr az arcon és a testen, férfi mintázatú eloszlással
- a szemöldök elváltozásai
- törékeny és hámló körmök
Ritka mellékhatások (1000 betegből legfeljebb 1 beteget érinthet):
- bőrpír vagy fájdalom a tenyérben vagy a talpban (tenyér-talpi eritrodiszesztézia szindróma)
Nagyon ritka mellékhatások (10 000 -ből legfeljebb 1 beteget érinthet)
- Szaruhártya -fekély vagy perforáció esetei
- Súlyos bőrreakciók, például hólyagosodás vagy hámlás (a Stevens-Johnson-szindrómát jelzi)
- A szem színes részének gyulladása
Mellékhatások bejelentése
Ha Önnél bármilyen mellékhatás jelentkezik, tájékoztassa erről kezelőorvosát vagy gyógyszerészét. Ez a betegtájékoztatóban fel nem sorolt bármilyen lehetséges mellékhatásra is vonatkozik. A mellékhatásokat közvetlenül a hatóság részére is bejelentheti az V. függelékben felsorolt jelentési rendszer segítségével. A mellékhatások bejelentésével Ön is hozzájárulhat ahhoz, hogy minél több információ álljon rendelkezésre a gyógyszer biztonságosságáról.
Lejárat és megőrzés
A gyógyszer gyermekektől elzárva tartandó!
A buborékcsomagoláson és a dobozon feltüntetett lejárati idő (EXP / EXP) után ne alkalmazza ezt a gyógyszert. A lejárati idő a hónap utolsó napjára vonatkozik.
Ez a gyógyszer nem igényel különleges tárolási körülményeket.
Semmilyen gyógyszert ne dobjon a szennyvízbe vagy a háztartási hulladékba. Kérdezze meg gyógyszerészét, hogy mit tegyen a már nem használt gyógyszereivel. Ez elősegíti a környezet védelmét.
Összetétel és gyógyszerforma
Mit tartalmaz a Tarceva?
- A Tarceva hatóanyaga az erlotinib. Minden filmtabletta 25 mg, 100 mg vagy 150 mg erlotinibet (erlotinib-hidroklorid formájában) tartalmaz, az erősségtől függően.
- Egyéb összetevők: Tablettamag: laktóz -monohidrát, mikrokristályos cellulóz, A típusú nátrium -keményítő -glikolát, nátrium -lauril -szulfát, magnézium -sztearát (laktóz -monohidrát tekintetében lásd még a 2. pontot). Tablettabevonat: hipromellóz, hidroxi -propil -cellulóz, titán -dioxid, makrogol.
Milyen a Tarceva külleme és mit tartalmaz a csomagolás?
A Tarceva 25 mg fehér vagy sárgás színű, kerek filmtabletta, egyik oldalán "T 25" bevéséssel, és 30 tablettát tartalmazó csomagolásban kapható.
A Tarceva 100 mg fehér vagy sárgás színű, kerek filmtabletta, egyik oldalán "T 100" bevéséssel, és 30 tablettát tartalmazó kiszerelésben kerül forgalomba.
A Tarceva 150 mg fehér vagy sárgás, kerek filmtabletta, egyik oldalán "T 150" bevéséssel, 30 tablettát tartalmazó kiszerelésben kerül forgalomba.
Forrás betegtájékoztató: AIFA (Olasz Gyógyszerügynökség). A tartalom 2016 januárjában jelent meg. A jelenlévő információk nem feltétlenül naprakészek.
A legfrissebb verzióhoz való hozzáféréshez ajánlatos az AIFA (Olasz Gyógyszerügynökség) webhelyét elérni. Jogi nyilatkozat és hasznos információk.
01.0 A GYÓGYSZER MEGNEVEZÉSE
FARMÁVAL BEVONATT TARCEVA 150 MG TABLETTA
02.0 MINŐSÉGI ÉS MENNYISÉGI ÖSSZETÉTEL
Egy filmtabletta 150 mg erlotinibet tartalmaz (erlotinib-hidroklorid formájában).
Ismert hatású segédanyagok: mindegyik filmtabletta 103,82 mg laktóz-monohidrátot tartalmaz.
A segédanyagok teljes listáját lásd a 6.1 pontban.
03.0 GYÓGYSZERFORMA
Filmtabletta.
Fehér vagy sárgás, kerek, mindkét oldalán domború tabletta, egyik oldalán "T 150" bevéséssel.
04.0 KLINIKAI INFORMÁCIÓK
04.1 Terápiás javallatok
Nem kissejtes tüdőkarcinóma ( Nem kissejtes tüdőrák (NSCLC):
A Tarceva lokálisan előrehaladott vagy áttétes nem kissejtes tüdőrákban (NSCLC) szenvedő betegek első vonalbeli kezelésére javallt, aktiváló EGFR-mutációkkal.
A Tarceva váltó fenntartó kezelésként javallott olyan betegeknél is, akik lokálisan előrehaladott vagy áttétes NSCLC-vel rendelkeznek, aktiváló EGFR-mutációkkal és stabil betegséggel az első vonalbeli kemoterápia után.
A Tarceva lokálisan előrehaladott vagy áttétes NSCLC -ben szenvedő betegek kezelésére is javallott, legalább egy korábbi kemoterápiás kezelés sikertelensége után.
A Tarceva felírásakor figyelembe kell venni a megnövekedett túléléshez kapcsolódó tényezőket.
A kezelés nem mutatott túlélési előnyöket vagy egyéb klinikailag releváns hatásokat epidermális növekedési faktor receptor (EGFR) -IHC negatív daganatban szenvedő betegeknél (lásd 5.1 pont).
Hasnyálmirigyrák:
A Tarceva gemcitabinnal kombinációban áttétes hasnyálmirigyrákos betegek kezelésére javallt.
A Tarceva felírásakor figyelembe kell venni a túléléshez kapcsolódó tényezőket (lásd 4.2 és 5.1 pont).
Lokálisan előrehaladott betegségben szenvedő betegeknél nem bizonyítottak túlélési előnyöket.
04.2 Adagolás és alkalmazás
A Tarceva -kezelést a daganatellenes kezelésekben jártas orvosnak kell felügyelnie.
Nem kissejtes tüdőkarcinómában szenvedő betegek:
EGFR-mutációs vizsgálatot kell végezni a Tarceva-kezelés megkezdése előtt olyan kemoterápiában nem részesült betegeknél, akik előrehaladott vagy áttétes NSCLC-ben szenvednek.
A Tarceva ajánlott napi adagja 150 mg, étkezés előtt legalább egy órával vagy azt követően két órával bevéve.
Hasnyálmirigyrákban szenvedő betegek:
A Tarceva ajánlott napi adagja 100 mg, amelyet legalább egy órával étkezés előtt vagy két órával étkezés előtt, gemcitabinnal kombinálva kell bevenni (lásd a gemcitabin alkalmazási előírását a hasnyálmirigyrák indikációjában).
Azon betegeknél, akiknél a kezelés első 4-8 hetében nem jelentkezik bőrkiütés, újra kell értékelni, hogy folytatni kell-e a Tarceva-kezelést (lásd 5.1 pont).
Ha az adagot módosítani kell, azt minden alkalommal 50 mg -mal kell csökkenteni (lásd 4.4 pont).
A Tarceva 25 mg, 100 mg és 150 mg hatáserősségben kapható.
A CYP3A4 szubsztrátok és modulátorok egyidejű alkalmazása szükségessé teheti az adagolás módosítását (lásd 4.5 pont).
Májelégtelenségben szenvedő betegek: Az erlotinib eliminációja a máj metabolizmusán és az epével történő kiválasztáson keresztül történik. Bár az erlotinib-expozíció hasonló volt mérsékelt májkárosodásban (Child-Pugh pontszám 7-9) és megfelelő májfunkciójú betegekben, óvatosan kell eljárni, amikor a Tarceva-t májkárosodásban szenvedő betegeknek adják. Ha súlyos mellékhatások jelentkeznek, meg kell fontolni a Tarceva -kezelés csökkentését vagy abbahagyását. Az erlotinib biztonságosságát és hatásosságát nem vizsgálták súlyos májelégtelenségben (AST / SGOT és ALT / SGPT> 5x ULN) szenvedő betegeknél. A Tarceva alkalmazása súlyos májelégtelenségben szenvedő betegeknél nem javasolt (lásd 5.2 pont).
Veseelégtelenségben szenvedő betegek: Az erlotinib biztonságosságát és hatásosságát nem vizsgálták veseelégtelenségben szenvedő betegeknél (a szérum kreatinin> a normál felső határának másfélszerese). A farmakokinetikai adatok alapján enyhe vagy közepesen súlyos veseelégtelenségben szenvedő betegeknél nem szükséges az adagolás megváltoztatása ( lásd a 5.2 pontot) A Tarceva alkalmazása súlyos veseelégtelenségben szenvedő betegeknél nem ajánlott.
Gyermekpopuláció: Az erlotinib biztonságosságát és hatásosságát 18 évesnél fiatalabb betegeknél nem igazolták A Tarceva alkalmazása gyermekeknél nem ajánlott.
Dohányosok: A cigaretta dohányzásról kimutatták, hogy 50-60%-kal csökkenti az erlotinib expozíciót. A Tarceva maximális tolerált dózisa NSCLC-s betegeknél, akik cigarettáznak, 300 mg volt. A kezdeti ajánlott adagnál magasabb hosszú távú hatékonyságot és biztonságosságot nem állapítottak meg betegeknél akik továbbra is dohányoznak (lásd 4.5 és 5.2 pont). Ezért a dohányosokat tanácsolni kell a dohányzás abbahagyására, mert az erlotinib plazmakoncentrációja a dohányosoknál alacsonyabb, mint a nemdohányzóknál.
04.3 Ellenjavallatok
Túlérzékenység az erlotinibre vagy a 6.1 pontban felsorolt bármely segédanyagra.
04.4 Különleges figyelmeztetések és a használathoz szükséges óvintézkedések
Az EGFR mutáció állapotának értékelése:
A páciens EGFR mutációs állapotának felméréséhez fontos, hogy jól validált és megbízható módszert válasszanak a hamis negatív vagy hamis pozitív meghatározások elkerülése érdekében.
Dohányosok
A dohányosokat tanácsolni kell a dohányzás abbahagyására, mert az erlotinib plazmakoncentrációja a dohányosoknál alacsonyabb, mint a nemdohányzóknál. A csökkenés mértéke klinikailag jelentős lehet (lásd 4.5 pont).
Tüdő intersticiális betegség (ILD))
Nem ritkán ILD-szerű, esetenként halálos kimenetelű eseményeket jelentettek azoknál a betegeknél, akik a Tarceva-t nem kissejtes tüdőrák (NSCLC), hasnyálmirigyrák vagy más előrehaladott szilárd daganatok kezelésére szedték. Az NSCLC-ben végzett kulcsfontosságú BR.21 vizsgálatban az ILD incidenciája (0,8%) azonos volt a placebo és a Tarceva csoportban. % a Tarceva és a gemcitabin csoportban, míg a placebo és a gemcitabin csoportban 0,4%. A Tarceva-val kezelt betegek összes előfordulása az összes vizsgálatban (beleértve a kontrollálatlan és az egyidejű kemoterápiát is) megközelítőleg 0,6%, szemben a placebóval kezelt betegek 0,2% -ával. Az ILD-szerű eseményekkel gyanús betegeknél jelentett diagnózisok között tüdőgyulladás, sugárzásos tüdőgyulladás, túlérzékenységi tüdőgyulladás, intersticiális tüdőgyulladás, tüdőközi intersticiális betegség, obliter bronchiolitis, tüdőfibrózis, akut légzési distressz szindróma, alveolitis és "tüdőinfiltráció". A tünetek a Tarceva-kezelés megkezdése után néhány napon vagy több hónapon belül jelentkeztek. Zavaró vagy egyidejű tényezők, például egyidejű vagy korábbi kemoterápia, korábbi sugárterápia, meglévő parenchymás tüdőbetegség, áttétek vagy tüdőfertőzések Az ILD gyakoribb előfordulása (kb. a halálozási arány 1,5%) volt megfigyelhető a japán származású lakosság körében.
Azoknál a betegeknél, akiknél akut új és / vagy progresszív, megmagyarázhatatlan tüdőtünetek, például dyspnoe, köhögés és láz jelentkeznek, a Tarceva -kezelést fel kell függeszteni a diagnosztikai értékelésig. Az erlotinibdel és a gemcitabinnal egyidejűleg kezelt betegeket gondosan ellenőrizni kell, hogy nincs-e ILD-szerű toxicitás. Ha ILD -t diagnosztizálnak, a Tarceva -kezelést abba kell hagyni, és szükség szerint megfelelő kezelést kell kezdeni (lásd 4.8 pont).
Hasmenés, kiszáradás, elektrolit -egyensúlyhiány és veseelégtelenség
A Tarceva -val kezelt betegek körülbelül 50% -ánál hasmenés jelentkezett (beleértve a nagyon ritka, halálos kimenetelű eseteket is); közepes vagy súlyos hasmenést kell kezelni, például loperamiddal. Bizonyos esetekben szükség lehet az adag csökkentésére. A klinikai vizsgálatok során az adagokat egyszerre 50 mg -ra csökkentették. Egyszeri 25 mg -os dóziscsökkentést nem vizsgáltak. A kiszáradással járó súlyos vagy tartós hasmenés, hányinger, étvágytalanság vagy hányás esetén a Tarceva beadását abba kell hagyni, és megfelelő intézkedéseket kell hozni a kiszáradás kezelésére (lásd 4.8 pont). Ritka esetekben hypokalaemiát és veseelégtelenséget jelentettek (beleértve a halálos eseteket is). Néhány eset másodlagos volt a hasmenés, hányás és / vagy étvágytalanság okozta súlyos kiszáradás miatt, míg mások az egyidejű kemoterápia miatt. Súlyosabb vagy tartós hasmenés vagy kiszáradáshoz vezető esetekben, különösen súlyosbító kockázati tényezőkkel rendelkező betegcsoportokban (különösen kemoterápiás kezeléssel és más gyógyszerekkel, tünetekkel vagy állapotokkal vagy egyéb hajlamosító feltételekkel együtt, beleértve a kor előrehaladtát is) a Tarceva -t be kell adni abba kell hagyni, és megfelelő intézkedéseket kell hozni a beteg intenzív intravénás rehidratálására.Ezenkívül a dehidráció kockázatának kitett betegeknél ellenőrizni kell a vesefunkciót és a szérum elektrolitjait, beleértve a káliumot is.
Hepatitis, májelégtelenség
A Tarceva -kezelés során ritka májelégtelenségről számoltak be (beleértve a halálos eseteket is). Zavaró tényezőnek tekintették a már meglévő májbetegséget vagy a hepatotoxikus gyógyszerek együttes alkalmazását. Ilyen betegeknél ezért fontolóra kell venni a májfunkció időszakos vizsgálatát. A Tarceva alkalmazását abba kell hagyni, ha a májfunkció rendellenességei súlyosak (lásd 4.8 pont). A Tarceva alkalmazása súlyos májelégtelenségben szenvedő betegeknél nem ajánlott.
Emésztőrendszeri perforáció
A Tarceva -t szedő betegeknél fokozott a gasztrointesztinális perforáció kialakulásának kockázata, amelyet ritkán figyelnek meg (beleértve néhány halálos esetet is). A kockázat magasabb azoknál a betegeknél, akik egyidejűleg antiangiogén szereket, kortikoszteroidokat, nem szteroid gyulladáscsökkentőket és / vagy taxán-alapú kemoterápiát szednek, vagy akiknek kórtörténetében peptikus fekély vagy divertikuláris betegség szerepel. A Tarceva -kezelést véglegesen fel kell függeszteni azoknál a betegeknél, akiknél gyomor -bélrendszeri perforáció alakul ki (lásd 4.8 pont).
Bullous, exfoliatív bőrbetegségek
Bullous, hólyagos és hámló bőrbetegségeket jelentettek, köztük nagyon ritka eseteket, amelyek Stevens-Johnson szindrómát / toxikus epidermális nekrolízist jeleznek, amelyek egyes esetekben halálosak voltak (lásd 4.8 pont). A Tarceva -kezelést meg kell szakítani vagy abba kell hagyni, ha a betegnél súlyos hólyagos, hólyagos vagy exfoliatív rendellenességek alakulnak ki. A hólyagos és hámló bőrváltozásokban szenvedő betegeket meg kell vizsgálni a bőrfertőzések szempontjából, és a helyi irányelvek szerint kell kezelni.
A szem patológiái
Azokat a betegeket, akiknél keratitisre utaló jelek vagy tünetek jelentkeznek, például akut szemgyulladás vagy a szem rosszabbodása, könnyezés, fotofóbia, homályos látás, szemfájdalom és / vagy vörös szem, haladéktalanul szemész szakorvoshoz kell utalni. Ha megerősítik a fekélyes keratitis diagnózisát, a Tarceva -kezelést fel kell függeszteni vagy abba kell hagyni. Keratitis diagnosztizálása esetén alaposan mérlegelni kell a kezelés folytatásának előnyeit és kockázatait. A Tarceva -t óvatosan kell alkalmazni olyan betegeknél, akiknek anamnézisében keratitis, fekélyes keratitis vagy súlyos szemszárazság áll fenn. A kontaktlencse használata a keratitis és a fekélyesedés kockázati tényezője is. Nagyon ritkán szaruhártya -perforációról vagy fekélyről számoltak be a Tarceva alkalmazása során (lásd 4.8 pont).
Kölcsönhatások más gyógyszerekkel
Az erős CYP3A4 -induktorok csökkenthetik az erlotinib hatékonyságát, míg az erős CYP3A4 -inhibitorok fokozott toxicitáshoz vezethetnek.
Az interakció egyéb formái
Az erlotinibet az oldhatóság csökkenése jellemzi 5 feletti pH -értékek esetén. Azok a gyógyszerek, amelyek megváltoztatják a gasztrointesztinális traktus (GI) felső részének pH -ját, mint például a protonpumpa -gátlók, a H2 -antagonisták és az antacidok, befolyásolhatják az erlotinib oldhatóságát, ezért annak biológiai hozzáférhetősége. A Tarceva adagjának növelése, ha ezeket a gyógyszereket egyidejűleg alkalmazzák, nem kompenzálja az expozíció csökkenését. Kerülni kell az erlotinib és a protonpumpa -gátlók kombinációját. Az erlotinib H2 -antagonistákkal és antacidokkal történő egyidejű alkalmazásának hatásai nem ismertek, azonban valószínű, hogy csökken a biológiai hozzáférhetőség. Ezért ezeket az egyidejű alkalmazást kerülni kell. Ha az antacidokat szükségesnek tartják a Tarceva -kezelés alatt, azokat legalább 4 órával a Tarceva napi adagja előtt vagy 2 órával azután kell bevenni.
A tabletták laktózt tartalmaznak, és nem adható olyan betegeknek, akik ritka, örökletes galaktóz intoleranciában, Lapp laktázhiányban vagy glükóz-galaktóz felszívódási zavarban szenvednek.
04.5 Kölcsönhatások más gyógyszerekkel és más interakciók
Interakciós vizsgálatokat csak felnőtteknél végeztek.
Erlotinib és más CYP szubsztrátok
Az erlotinib a CYP1A1 hatékony inhibitora, a CYP3A4 és a CYP2C8 mérsékelt inhibitora, valamint a glükuronidáció erős inhibitora in vitro az UGT1A1 által.
A CYP1A1 emberi szövetekben való nagyon csökkent expressziója miatt a CYP1A1 erős gátlásának élettani jelentősége nem ismert.
Amikor az erlotinibet ciprofloxacinnal, mérsékelt CYP1A2-gátlóval együtt adták, az erlotinib-expozíció [AUC] szignifikánsan, 39% -kal emelkedett, míg a Cmax-ban nem észleltek statisztikailag szignifikáns változást. Az aktív metabolit expozíció kb. AUC, ill. csökkenthető.
A Tarceva előkezelése vagy egyidejű alkalmazása nem változtatta meg a CYP3A4 prototípusos szubsztrátok, például a midazolám és az eritromicin clearance-ét, de úgy tűnt, hogy akár 24%-kal csökkenti a midazolám orális biohasznosulását. Egy másik klinikai vizsgálatban az erlotinib nem változtatta meg a CYP3A4 / 2C8 egyidejűleg alkalmazott paklitaxel szubsztrátjának farmakokinetikáját. Ezért nem valószínű, hogy jelentős kölcsönhatások lépnének fel más CYP3A4 szubsztrátok clearance -eivel.
A glükuronidáció gátlása kölcsönhatásokat okozhat olyan gyógyszerekkel, amelyek az UGT1A1 szubsztrátjai, és amelyek kizárólag ezen az úton ürülnek ki. Azoknál a betegeknél, akiknél csökkent az UGT1A1 expresszió szintje vagy a glükuronidáció genetikai elváltozásai (pl. Gilbert -kór), megemelkedhet a szérum bilirubin koncentrációja, és óvatosan kell őket kezelni.
Emberben az erlotinib a májban a máj citokrómjai által metabolizálódik, főként a CYP3A4 és kisebb mértékben a CYP1A2 által. Az extrahepatikus metabolizmus, amelyet a bélben a CYP3A4, a tüdőben a CYP1A1 és a daganatszövetben közvetít, szintén hozzájárulhat az erlotinib metabolikus clearance -e. Lehetséges kölcsönhatások a hatóanyagokkal, amelyeket ezek az enzimek metabolizálnak, vagy amelyek gátlóként vagy induktorokként hatnak rájuk.
A CYP3A4 aktivitás erős inhibitorai csökkentik az erlotinib anyagcseréjét és növelik az erlotinib plazmakoncentrációját. Egy klinikai vizsgálatban az erlotinib és a ketokonazol (napi kétszer 200 mg, naponta kétszer, 5 napon keresztül), egy erős CYP3A4 inhibitor egyidejű alkalmazása növelte az erlotinib expozíció (az AUC 86% -a és a Cmax 69% -a). Ezért óvatosan kell eljárni, ha erlotinibet adnak egy erős CYP3A4 -gátlóval, például azol gombaellenes szerekkel (pl. Ketokonazol, itrakonazol, vorikonazol), proteázgátlókkal, eritromicinnel vagy klaritromicinnel együtt, erlotinib adaggal, különösen toxicitás esetén.
Erős CYP3A4 -induktorok fokozzák az erlotinib metabolizmusát, és jelentősen csökkentik az erlotinib plazmakoncentrációját. Egy klinikai vizsgálatban az erlotinib és a rifampicin (600 mg / nap, szájon át 7 napon keresztül), a CYP3A4 erős induktora egyidejű alkalmazása 69% -kal csökkent az erlotinib átlagos AUC-értéke. A rifampicin és egyetlen 450 mg-os Tarceva adag együttes alkalmazása 57,5% -os átlagos erlotinib-expozíciót (AUC) eredményezett ahhoz képest, mint amennyit egyetlen 150 mg-os Tarceva adag beadása után elértünk. a rifampicin -kezelés hiánya. Ezért kerülni kell a Tarceva és a CYP3A4 induktorok együttes alkalmazását. Azoknál a betegeknél, akik Tarceva -val és egy erős CYP3A4 -induktorral, például rifampicinnel egyidejűleg kezelést igényelnek, mérlegelni kell az adag 300 mg -ra történő emelését, miközben biztonságosságukat (beleértve a vese- és májfunkciót és a szérum elektrolitokat) gondosan figyelemmel kell kísérni, és ha jól tolerálják több 2 hétnél hosszabb ideig, az adag további 450 mg -ra történő emelését lehet fontolóra venni szoros biztonsági ellenőrzés mellett. Az expozíció csökkenése más induktorokkal, például fenitoinnal, karbamazepinnel, barbiturátokkal vagy orbáncfűvel is előfordulhat.Hypericum perforatum). Óvatosan kell eljárni, ha ezeket a hatóanyagokat erlotinibdel kombinálják. Amikor csak lehetséges, mérlegelni kell az alternatív kezeléseket, amelyekben nincs erős induktív CYP3A4 -aktivitás.
Erlotinib és kumarin eredetű antikoagulánsok
A Tarceva-t kapó betegeknél beszámoltak a kumarinból származó antikoagulánsokkal, köztük a warfarinnal való kölcsönhatásokról, amelyek megnövekedett INR-t (International Normalized Ratio) és vérzéses eseményeket eredményeztek, amelyek egyes esetekben halálos kimenetelűek voltak. bármilyen változás a protrombin időben vagy az INR -ben.
Erlotinib és sztatinok
A Tarceva és egy sztatin kombinációja növelheti a sztatin által kiváltott myopathia kockázatát, beleértve a rabdomiolízist is, amelyet ritkán figyeltek meg.
Erlotinib és dohányosok
Egy farmakokinetikai interakciós vizsgálat eredményei a Tarceva beadása után szignifikáns, 2,8 -as csökkenést mutattak; A dohányzókban az AUCinf, a Cmax és a 24 órás plazmakoncentráció 1,5 és 9-szerese a nemdohányzókhoz képest (lásd 5.2 pont). Ezért a még dohányzó betegeket arra kell ösztönözni, hogy mielőbb hagyják abba a dohányzást. a Tarceva -kezelés megkezdése, különben az erlotinib plazmakoncentrációja csökken. A csökkent expozíció klinikai hatását nem állapították meg véglegesen, de klinikailag jelentős lehet.
Erlotinib és P-glikoprotein inhibitorok
Az erlotinib a P-glikoprotein szubsztrátja, a hatóanyag hordozója. A P-glikoprotein inhibitorok, például ciklosporin és verapamil együttes alkalmazása megváltoztathatja az erlotinib eloszlását és / vagy eliminációját. Ennek a kölcsönhatásnak a következményeit, például a központi idegrendszerre gyakorolt toxicitást, nem állapították meg. Az ilyen helyzetekben óvatosan kell eljárni.
Erlotinib és a pH -t megváltoztató gyógyszerek
Az erlotinibet az oldhatóság csökkenése jellemzi 5 feletti pH -értékeknél. Azok a gyógyszerek, amelyek megváltoztatják a gasztrointesztinális traktus (GI) felső részének pH -ját, megváltoztathatják az erlotinib oldhatóságát és következésképpen biológiai hozzáférhetőségét. Az erlotinib és a protonpumpa-gátló (PPI) omeprazol együttes alkalmazása 46% -kal, illetve 61% -kal csökkentette az erlotinib-expozíciót [AUC] és a csúcskoncentrációt [Cmax].A Tmax vagy a felezési idő változását nem észlelték: A Tarceva 300 mg ranitidinnel, H2-receptor-antagonistával történő egyidejű alkalmazása 33 % -kal, 54 % -kal csökkentette az erlotinib-expozíciót [AUC] és a csúcskoncentrációt [Cmax]. A Tarceva adagjának növelése, ha ezeket a gyógyszereket egyidejűleg alkalmazzák, nem kompenzálja az expozíció csökkenését. Ha azonban a Tarceva -t 2 órával az adagolás előtt 2 órával vagy 10 órával az adagolás után kétszer adták be, az erlotinib -expozíció [AUC] és a csúcskoncentráció [Cmax] csak 15% -kal, illetve 17% -kal csökkent. Az antacidok hatása az erlotinib felszívódására nem vizsgálták, de a felszívódás romolhat, ami alacsonyabb plazmaszintet eredményezhet. Összefoglalva, el kell kerülni az erlotinib és a protonpumpa -gátlók kombinációját. Ha megfontoljuk a ranitidin alkalmazását, akkor a két gyógyszert szakaszosan kell beadni: A Tarceva -t legalább 2 órával a ranitidin beadása előtt vagy 10 órával kell bevenni.
Erlotinib és Gemcitabine
Egy fázis Ib vizsgálatban a gemcitabinnak nem volt jelentős hatása az erlotinib farmakokinetikájára, és az erlotinibnek sem volt jelentős hatása a gemcitabin farmakokinetikájára.
Erlotinib és karboplatin / paclitaxel
Az erlotinib növeli a platina koncentrációt. Egy klinikai vizsgálatban az erlotinib karboplatinnal és paklitaxellel történő együttes alkalmazása a teljes platina AUC0-48 értékét 10,6%-kal növelte. Bár ez a növekedés statisztikailag szignifikáns, ennek a különbségnek a nagysága nem tekinthető klinikailag relevánsnak. A klinikai gyakorlatban előfordulhatnak más társtényezők is, amelyek a karboplatin-expozíció fokozódásához vezethetnek, például veseelégtelenség. A karboplatin ill. paklitaxel az erlotinib farmakokinetikájára vonatkozóan.
Erlotinib és kapecitabin
A kapecitabin növelheti az erlotinib koncentrációját. Amikor az erlotinibet kapecitabinnal együtt adták, statisztikailag szignifikánsan nőtt az erlotinib AUC és kismértékben a Cmax, összehasonlítva egy másik vizsgálatban megfigyelt értékekkel, amelyben az erlotinibet önmagában alkalmazták. Az erlotinibnek nincs jelentős hatása a farmakokinetikára kapecitabin.
Erlotinib és proteaszóma inhibitorok
Ami a hatásmechanizmust illeti, a proteasóma -inhibitorok, beleértve a bortezomibot is, befolyásolhatják az EGFR -inhibitorok, köztük az erlotinib aktivitását. Ezt a hatást alátámasztja az EGFR proteaszómán keresztüli lebomlását kiemelő klinikai és preklinikai adatok korlátozott rendelkezésre állása.
04.6 Terhesség és szoptatás
Terhesség
Nincsenek megfelelő adatok az erlotinib terhes nőkön történő alkalmazására. Állatkísérletek nem mutattak ki teratogenitást vagy rendellenes születést. A terhességre gyakorolt negatív hatást azonban nem lehet kizárni, mivel patkányokon és nyulakon végzett vizsgálatok fokozott embrió / magzati halálozást mutattak (lásd 5.3. szakasz) Az emberekre gyakorolt lehetséges kockázat nem ismert.
Fogamzóképes korú nők
A fogamzóképes nőket tanácsolni kell, hogy kerüljék a terhességet a Tarceva -kezelés alatt. Megfelelő fogamzásgátló módszereket kell alkalmazni a kezelés alatt és a kezelés befejezése után legalább 2 hétig. Terhes nőknél a kezelést csak akkor szabad folytatni, ha az anya számára várható előny meghaladja a magzatra gyakorolt kockázatot.
Etetési idő
Nem ismert, hogy az erlotinib kiválasztódik -e az anyatejbe. Az újszülött esetleges károsodása miatt az anyákat tanácsolni kell, hogy ne szoptassanak a Tarceva -kezelés alatt.
Termékenység
Állatkísérletek nem mutattak ki károsodott termékenységet. Mindazonáltal nem zárható ki a termékenységre gyakorolt negatív hatás, mivel állatkísérletek kimutatták a reproduktív paraméterekre gyakorolt hatást (lásd 5.3 pont). Az emberekre gyakorolt lehetséges kockázat nem ismert.
04.7 Hatások a gépjárművezetéshez és gépek kezeléséhez szükséges képességekre
A gépjárművezetéshez és gépek kezeléséhez szükséges képességeket nem vizsgálták, azonban az erlotinib nem jár károsodott mentális képességekkel.
04.8 Nemkívánatos hatások
Nem kissejtes tüdőrák (Tarceva önmagában):
Egy randomizált kettős vak vizsgálatban (BR.21; Tarceva második vonalbeli terápiaként) a leggyakrabban jelentett mellékhatások a bőrkiütés (75%) és a hasmenés (54%) voltak, a legtöbb esetben az 1. fokozatú intenzitással. /2 és kezelhető minden beavatkozás nélkül. A 3/4 fokozatú kiütés és hasmenés a Tarceva -val kezelt betegek 9% -ánál, illetve 6% -ánál fordult elő, és mindkettő miatt a vizsgálatot a betegek 1% -ánál le kellett állítani, az adagolás a betegek 6% -ánál, illetve 1% -a volt. A BR.21 vizsgálatban a kiütés megjelenéséig eltelt medián idő 8 nap, a hasmenés megjelenéséig eltelt idő pedig 12 nap volt.
Általánosságban elmondható, hogy a kiütés "enyhe vagy mérsékelt erythematosus és papularis-pustuláris kitörésként nyilvánul meg, amely a napsugárzásnak kitett területeken előfordulhat vagy súlyosbodhat. A napnak kitett betegek számára tanácsos védőruházatot használni" és / vagy fényvédők (pl. ásványi anyagok alapján).
Az 1. táblázat az NCI-CTC besorolása (National Cancer Institute Common Toxicity Criteria) szerint foglalja össze azokat a mellékhatásokat, amelyek gyakrabban (≥3%) fordultak elő a Tarceva-val kezelt betegek körében, összehasonlítva a BR.21 és a a Tarceva csoportba tartozó betegek legalább 10% -a.
A következő kifejezéseket használják a nemkívánatos hatások gyakoriság szerinti osztályozására: nagyon gyakori (≥1 / 10); gyakori (≥1 / 100,
Az egyes gyakorisági csoportokon belül a mellékhatásokat csökkenő súlyosság szerint sorolják fel.
1. táblázat: Nagyon gyakori mellékhatások a BR.21 vizsgálatban
* Súlyos fertőzések, neutropeniával vagy anélkül, beleértve a tüdőgyulladást, a szepszist és a cellulitist.
** Dehidratációhoz, hypokalaemiához és veseelégtelenséghez vezethet.
*** A kiütések között pattanásos bőrgyulladás is szerepelt.
2 másik randomizált, kettős vak, placebo-kontrollos fázis III vizsgálatban: BO18192, (SATURN) és BO25460 (IUNO); A Tarceva-t fenntartó terápiaként adták az első vonalbeli kemoterápia után. Ezeket a vizsgálatokat összesen 1532, előrehaladott, visszaeső vagy áttétes NSCLC-ben szenvedő, standard elsővonalbeli platina-alapú kemoterápiát követő betegben végezték, új biztonsági jelentést nem azonosítottak.
A BO18192 és BO25460 vizsgálatokban a Tarceva-val kezelt betegeknél leggyakrabban megfigyelt mellékhatások a kiütés és a hasmenés voltak (lásd 2. táblázat). Egyik vizsgálatban sem figyeltek meg 4. fokú kiütést vagy hasmenést. Kiütés és hasmenés miatt a Tarceva -kezelést 1% -ban, illetve 1% -ban hagyták abba
2. táblázat: A BO18192 (SATURN) és a BO25460 (IUNO) vizsgálatok leggyakoribb mellékhatásai
* Biztonsági Értékelő Csoport
Egy nyílt, III. Fázisú vizsgálatban, ML 20650, 154 betegnél, a Tarceva biztonságosságát NSCLC és EGFR aktiváló mutációkban szenvedő betegek első vonalbeli kezelésében 75 betegnél értékelték; nem találtak új kapcsolódó jelentéseket.
A Tarceva-val kezelt betegeknél a leggyakrabban megfigyelt mellékhatások az ML 20650-ben voltak kiütések és hasmenés (bármilyen fokozatú, 80% és 57%), többnyire 1. és 2. fokú és kezelhető. 3. fokozatú kiütés és hasmenés 9% -ban és 4% -ban fordult elő A betegek % -a, illetve 4. fokú kiütés vagy hasmenés. A kiütések és hasmenések miatt a Tarceva -kezelést a betegek 1 % -ánál abba kellett hagyni. Kiütés és hasmenés miatti dózismódosításra (megszakításra vagy csökkentésre) a betegek 11% -án, illetve 7% -án volt szükség.
Hasnyálmirigyrák (Tarceva gemcitabinnal egyidejűleg):
A PA.3 kulcsfontosságú vizsgálatban a leggyakoribb mellékhatások 100 mg Tarceva -val és gemcitabinnel kezelt hasnyálmirigyrákos betegeknél a fáradtság, kiütés és hasmenés voltak. A Tarceva plusz gemcitabin karon a betegek 5% -ánál jelentettek kiütést és 3/4 fokozatú hasmenést. A kiütés és a hasmenés megjelenéséig eltelt medián idő 10 nap, illetve 15 nap volt. A kiütések és a hasmenés a betegek 2% -ánál csökkentette az adagot, és a vizsgálatot abbahagyta a Tarceva -t és gemcitabint kapó betegek legfeljebb 1% -ánál.
A PA.3 kulcsfontosságú vizsgálatban a 100 mg Tarceva plusz gemcitabinnel kezelt betegeknél gyakrabban (≥3%) előforduló mellékhatások fordultak elő, mint a placebo + gemcitabin csoportban, és a 100 mg plusz Tarceva csoportba tartozó betegek legalább 10% -ában A gemcitabint a 3. táblázatban foglaltuk össze, a National Cancer Institute Common Toxicity Criteria (NCI-CTC) alapján.
A következő kifejezéseket használják a nemkívánatos hatások gyakoriság szerinti osztályozására: nagyon gyakori (≥1 / 10); gyakori (≥1 / 100,
Az egyes gyakorisági csoportokon belül a mellékhatásokat csökkenő súlyosság szerint sorolják fel.
3. táblázat: Nagyon gyakori mellékhatások a PA.3 vizsgálatban (100 mg kohort)
* Súlyos fertőzések, neutropeniával vagy anélkül, beleértve a tüdőgyulladást, a szepszist és a cellulitist.
** Dehidratációhoz, hypokalaemiához és veseelégtelenséghez vezethet.
*** A kiütések között pattanásos bőrgyulladás is szerepelt.
Egyéb megjegyzések:
A Tarceva biztonságossági értékelése több mint 1500 beteg adatain alapul, akiket legalább egy 150 mg -os Tarceva -dózissal kezeltek, és több mint 300 beteget kezeltek 100 vagy 150 mg Tarceva -val gemcitabinnal kombinálva.
A következő mellékhatásokat figyelték meg a Tarceva monoterápiával kezelt betegeknél és a Tarceva -val egyidejűleg kemoterápiával kezelt betegeknél.
A BR 21 és PA 3 vizsgálatok nagyon gyakori mellékhatásait az 1. és 3. táblázat tartalmazza, az egyéb mellékhatásokat, beleértve a más vizsgálatokból származó mellékhatásokat, a 4. táblázat foglalja össze.
Az egyes gyakorisági csoportokon belül a mellékhatásokat csökkenő súlyosság szerint sorolják fel.
4. táblázat: Mellékhatások gyakorisági kategóriák szerint
1 A PA.3 tanulmányban.
2 Beleértve a belül növekvő szempillákat, a túlzott növekedést és a szempillák megvastagodását.
3 Beleértve a halálos eseteket is, azoknál a betegeknél, akik Tarceva -t NSCLC vagy más előrehaladott szilárd daganatok kezelésére szedtek (lásd 4.4 pont). Japán származású betegeknél magasabb incidenciát figyeltek meg.
4 A klinikai vizsgálatok során egyes esetek összefüggésben álltak a warfarin egyidejű alkalmazásával (lásd 4.5 pont), és néha a nem szteroid gyulladáscsökkentők egyidejű alkalmazásával.
5 Beleértve az alanin -aminotranszferáz (ALT), az aszpartát -aminotranszferáz (AST) és a bilirubin emelkedését). Ezek leggyakrabban enyhe vagy mérsékelt jellegűek voltak, átmeneti jellegűek vagy máj áttétekhez kapcsolódtak.
6 Beleértve a halálos eseteket is. Zavaró tényezőnek tekintették a már meglévő májbetegséget vagy a hepatotoxikus gyógyszerek együttes alkalmazását (lásd 4.4 pont).
7 Beleértve a halálos eseteket is (lásd 4.4 pont).
A feltételezett mellékhatások bejelentése
A gyógyszer engedélyezése után jelentkező feltételezett mellékhatások bejelentése fontos, mivel lehetővé teszi a gyógyszer előny / kockázat arányának folyamatos nyomon követését. Az egészségügyi szakembereket kérjük, hogy jelentsék be a feltételezett mellékhatásokat a nemzeti jelentési rendszeren keresztül. "Address https: //www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Túladagolás
Tünetek
A Tarceva egyszeri, szájon át adott dózisait egészséges alanyoknál legfeljebb 1000 mg -ig, daganatos betegeknél pedig 1600 mg -ig tűrték. A napi kétszeri 200 mg -os ismételt adagokat egészséges személyeknél néhány napos alkalmazás után rosszul tolerálták. Ezekből a vizsgálatokból származó adatok alapján lehetséges, hogy az ajánlottnál nagyobb dózisok esetén súlyos mellékhatások, például hasmenés, kiütés és esetleg fokozott májaminotranszferáz -aktivitás léphetnek fel.
Menedzsment
Túladagolás gyanúja esetén a Tarceva alkalmazását abba kell hagyni, és tüneti kezelést kell kezdeni.
05.0 FARMAKOLÓGIAI TULAJDONSÁGOK
05.1 Farmakodinámiás tulajdonságok
Farmakoterápiás csoport: protein kináz inhibitor daganatellenes gyógyszer.
ATC kód: L01XE03.
A cselekvés mechanizmusa
Az erlotinib egy epidermális növekedési faktor receptor / humán epidermális növekedési faktor receptor (EGFR, más néven HER1) tirozin -kináz inhibitor. Az erlotinib az EGFR intracelluláris foszforilációjának hatékony inhibitora. Az EGFR a normál és a tumorsejtek sejtfelszínén expresszálódik, nem klinikai modellekben az EGFR foszfotirozin gátlása sejtmegállást és / vagy halált okoz.
Az EGFR mutációk "az anti-apoptotikus és proliferatív jelátviteli utak konstitutív aktiválódását eredményezhetik. Az erlotinib erőteljes hatékonysága az EGFR által közvetített jelátvitel gátlásában ezekben az EGFR mutáció-pozitív daganatokban az erlotinib és az ATP kötőhely közötti szoros kapcsolatnak tulajdonítható Az EGFR mutált kináz doménjében. A downstream transzdukciós jel blokkolása miatt a sejtproliferáció megáll, és a sejtek elhalása indukálódik a belső apoptotikus útvonalon keresztül. A tumor regressziója megfigyelhető. az egérmodellekben, amelyek ezen aktiváló EGFR mutációk markáns kifejeződését mutatják.
Klinikai hatékonyság
• A nem kissejtes tüdőrák (NSCLC) első vonalbeli terápiája aktiváló EGFR mutációjú betegeknél (Tarceva önmagában):
A Tarceva hatékonyságát az első vonalbeli betegeknél, akik aktiválták az EGFR-mutációkat az NSCLC-ben, egy randomizált, nyílt III. Fázisú vizsgálatban (ML20650, EURTAC) bizonyították. Ezt a vizsgálatot metasztatikus vagy lokálisan előrehaladott NSCLC -vel (IIIB és IV stádium) rendelkező kaukázusi betegekben végezték, akik korábban nem kaptak kemoterápiát vagy bármilyen szisztémás daganatellenes terápiát betegségük miatt, és mutációik voltak az EGFR tirozin -kináz doménjében (exon deléció). vagy exon 21 mutáció) A betegeket 1: 1 arányban randomizálták, hogy naponta 150 mg Tarceva-t vagy legfeljebb 4 ciklus platinaalapú fogszabályozó kemoterápiát kapjanak.
Az elsődleges végpont a kutató által értékelt PFS volt.
A hatékonysági eredményeket az 5. táblázat foglalja össze.
5. táblázat: A Tarceva hatékonysági eredményei a kemoterápiával szemben az ML20650 vizsgálatban (EURTAC)
CR = teljes válasz; RP = részleges válasz.
* A betegség előrehaladásának vagy halálának kockázatát 58% -kal csökkentették.
** A vizsgáló és az IRC PFS értékelése közötti általános egyetértési arány 70%volt.
*** "Magas kereszteződési arányt" figyeltek meg a kemoterápiás betegek 82% -ánál, akik ezt követően EGFR-hez kapcsolódó tirozin-kináz inhibitorral kezelték, és 2 kivételével minden beteget Tarceva-val kezeltek.
- NSCLC fenntartó terápia első vonalbeli kemoterápia után (Tarceva monoterápiaként):
A Tarceva hatékonyságát és biztonságosságát fenntartó terápiaként az NSCLC első vonalbeli kemoterápiája után egy randomizált, kettős-vak, placebo-kontrollos vizsgálatban (BO18192, SATURN) vizsgálták. Ebben a vizsgálatban 889 olyan lokálisan előrehaladott NSCLC-s beteg vett részt, akik nem haladtak előre 4 ciklus kétszeres, platinaalapú kemoterápia. A betegeket 1: 1 arányban randomizálták 150 mg Tarceva-val vagy placebóval szájon át naponta egyszer a progresszióig. L "A vizsgálat fő végpontja a progressziómentes túlélés (PFS) volt betegek. A demográfiai és betegségjellemzők a belépéskor jól kiegyensúlyozottak voltak a két kezelési ág között.Az ECOG PS> 1, jelentős máj- vagy vesebetegségben szenvedő betegeket nem vonták be a vizsgálatba.
Ebben a vizsgálatban a teljes populáció előnyös volt a PFS elsődleges végpont szempontjából (HR = 0,71p)
Az EGFR mutáció pozitív alcsoportjában a placebóval kezelt betegek 67% -a kapott EGFR-TKI inhibitorokat a második vagy az azt követő kezelési sorokban.
A BO25460 vizsgálatot (IUNO) 643 olyan betegben végezték, akiknek előrehaladott NSCLC-je volt, anélkül, hogy aktiválták volna az EGFR mutációkat (exon 19 deléció vagy 21 exon L858R mutáció), és akik nem mutatták ki a betegség progresszióját négy platinaalapú kemoterápia után.
A vizsgálat célja az volt, hogy összehasonlítsa a fenntartó első sorban adott erlotinib teljes túlélését a betegség progressziójának idején alkalmazott erlotinibhez képest. A vizsgálat nem felelt meg az elsődleges végpontnak. A Tarceva operációs rendszere a fenntartás során nem volt jobb, mint a Tarceva második vonalbeli kezelésénél, azoknál a betegeknél, akiknek a tumorában nem volt aktiváló EGFR-mutáció (HR = 1,02, 95% CI, 0,85-1, 22, p = 0,82). a progressziómentes túlélés (PFS) másodlagos végpontja nem különbözött a Tarceva és a placebo között a fenntartó kezelés során (HR = 0,94, 95% CI, 0,80-1,11; p = 0,48).
A BO25460 (IUNO) vizsgálat adatai alapján a Tarceva alkalmazása nem javasolt első vonalbeli fenntartó kezelésként olyan betegeknél, akiknél nincs aktiváló EGFR-mutáció.
- NSCLC kezelése legalább egy korábbi kemoterápiás sor sikertelensége után (Tarceva önmagában):
A Tarceva hatékonyságát és biztonságosságát, mint második / harmadik vonalbeli terápiát, egy randomizált, kettős vak, placebo-kontrollos vizsgálatban (BR.21) igazolták 731, lokálisan előrehaladott vagy áttétes NSCLC-ben szenvedő betegen, legalább egy kemoterápiás kezelés sikertelensége után. 2: 1 arányban randomizáltak 150 mg Tarceva-val vagy placebóval szájon át naponta egyszer. és fájdalom) és a biztonság Az elsődleges végpont a túlélés volt.
A demográfiai jellemzők jól kiegyensúlyozottak voltak a két kezelési csoport között. A betegek körülbelül kétharmada férfi volt, körülbelül egyharmaduk ECOG teljesítménnyel (PS) rendelkezett a felvételkor 2, illetve 9% -a ECOG PS-vel a felvételkor. Az összes beteg 93% -a és 92% -a. A Tarceva-csoportot és a placebo-csoportot korábban platina-alapú kezeléssel kezelték, és az összes beteg 36% -át, illetve 37% -át korábban taxánnal kezelték.
A korrigált kockázati arány (HR) a halálozásoknál a Tarceva csoportban a placebo csoporthoz képest 0,73 (95% CI: 0,60-0,87) (p = 0,001). A Tarceva -ban és a placebo -csoportban élő betegek 31,2% -a, illetve 21,5% -a volt életben 12 hónapos korban. A teljes túlélés medián értéke 6,7 hónap volt a Tarceva csoportban (95% CI: 5,5-7,8 hónap), míg a placebo csoportban 4,7 hónap (95% CI: 4,1-6, 3 hónap).
A teljes túlélésre gyakorolt hatást különböző betegcsoportokban vizsgálták. A Tarceva hatása a teljes túlélésre hasonló volt azoknál a betegeknél, akiknek kiindulási teljesítménye (ECOG) 2-3 volt (HR = 0,77, CI 95 % 0,6-1,0) vagy 0- 1 (HR = 0,73, 95% CI 0,6-0,9), férfiaknál (HR = 0,76, 95% CI 0, 6-0,9) vagy nőknél (HR = 0,80, 95% CI 0,6-1,1), kevesebb mint 65 éves (HR = 0,75, 95% CI 0,6-0,9) vagy idősebb betegeknél (HR = 0,79, 95% CI 0,6-1,0), korábbi kezeléssel kezelt betegeknél (HR = 0,76, 95% CI% 0,6) -1,0) vagy több korábbi kezeléssel (HR = 0,75, 95% CI 0,6-1,0), kaukázusi betegeknél (HR = 0,79, 95% CI 0,6-1,0) vagy ázsiaiakon (HR = 0,61, 95% CI 0,4-) 1,0), adenokarcinómában (HR = 0,71, 95% CI 0,6-0, 9) vagy pikkelysömörben (HR = 0,67, 95% CI 0,5-0,9) szenvedő betegeknél, de nem más szövettani betegeknél (HR 1,04, 95) % CI 0,7-1,5), IV. Stádiumú betegségben szenvedő betegeknél diagnózisnál (HR = 0,92, 95% CI 0,7-1,2) vagy stádiumban
Az ismert EGFR-expressziós állapotú betegek 45% -ánál a kockázati arány 0,68 (95% CI 0,49-0,94) volt az EGFR-pozitív daganatban szenvedő betegeknél és 0, 93 (95% CI 0,63-1,36) az EGFR-negatív daganatos betegeknél (az IHC határozza meg, az EGFR pharmDx készletet használva, mint EGFR negatív, 10% -nál kevesebb tumorsejt -jelöléssel rendelkezők). Az ismeretlen EGFR-expressziós állapotú betegek fennmaradó 55% -ánál a kockázati arány 0,77 volt (95% CI 0,61-0,98).
A medián PFS 9,7 hét volt a Tarceva csoportban (95% CI, 8,4-12,4 hét), míg a placebo csoportban 8,0 hét (95% CI, 7,9-8,1 hét).).
A Tarceva csoportban a RECIST objektív válaszarány 8,9% volt (95% CI, 6,4-12,0). Az első 330 beteget központilag értékelték (válaszarány 6, 2%); 401 beteget vizsgáltak (válaszarány 11,2%) ).
A válasz átlagos időtartama 34,3 hét volt, minimum 9,7 és maximum 57,6+ hét. A betegek 44,0% -a ért el teljes, részleges választ vagy betegség stabilizálódást a Tarceva -csoportban, míg a placebo -csoport 27,5% -a (p = 0,004).
A Tarceva túlélési előnyeit azoknál a betegeknél is észlelték, akik nem értek el objektív tumorválaszt (RECIST kritériumok). Ezt bizonyította a halálozási kockázati arány 0,82 (95% CI, 0,68-0,99) azon betegek körében, akik a betegség stabilizálódását vagy progresszióját érték el a legjobb válaszként.
A Tarceva tüneti előnyökkel járt, mivel jelentősen meghosszabbította a köhögés, a nehézlégzés és a fájdalom súlyosbodásának idejét a placebóhoz képest.
- Hasnyálmirigyrák (a Tarceva gemcitabinnel együtt adva a PA.3 vizsgálatban):
A Tarceva hatásosságát és biztonságosságát elsővonalbeli kezelésként gemcitabinnal kombinálva egy randomizált, kettős vak, placebo-kontrollos vizsgálatban értékelték, lokálisan előrehaladott, nem eltávolítható vagy áttétes hasnyálmirigyrákos betegekben. naponta folyamatosan és iv. gemcitabin (1000 mg / m2, 1. ciklus - 8. hét, 1., 8., 15., 22., 29., 36. és 43. nap; 2. és azt követő ciklusok - 1., 8. és 15. nap) 4 hetes ciklus [a hasnyálmirigyrák jóváhagyott adagja és ütemterve, lásd a gemcitabin alkalmazási előírását]). A Tarceva -t vagy a placebót naponta egyszer szájon át alkalmazták a betegség progressziójáig vagy az elfogadhatatlan toxicitásig. Az elsődleges végpont a teljes túlélés volt.
A betegek demográfiai és betegségjellemzői a belépéskor hasonlóak voltak a 2 kezelési csoportban, a Tarceva 100 mg plusz gemcitabin vagy a placebo plusz gemcitabin csoportban, kivéve az erlotinib / gemcitabin karban lévő nők valamivel magasabb arányát a placebo / gemcitabin karhoz képest:
A túlélést a kezelésre szánt populációban értékelték a követési túlélési adatok alapján. Az eredményeket a következő táblázat írja le (az áttétes és lokálisan előrehaladott betegségben szenvedő betegek csoportjaira vonatkozó eredmények egy feltáró alcsoport -elemzésből származnak).
Azok a betegek, akiknek a kiindulási időpontjában kedvező klinikai állapotuk volt (alacsony fájdalomintenzitás, jó életminőség és jó PS), jobban részesülhetnek a Tarceva-ból, amint azt egy "post-hoc elemzés" is mutatja. Az előny elsősorban az alacsony fájdalomintenzitás jelenlétéből származik. .
Egy poszt-hoc elemzés szerint a Tarceva-val kezelt betegeknél, akiknél kiütés alakult ki, hosszabb volt az általános túlélés, mint azoknál a betegeknél, akiknél nem jelentkezett kiütés (átlagos OS 7,2 hónap kontra 5 hónap, HR: 0,61).
A Tarceva -t szedő betegek 90% -ánál kiütés alakult ki az első 44 napon belül. A kiütés megjelenéséig eltelt medián idő 10 nap volt.
Gyermekpopuláció
Az Európai Gyógyszerügynökség eltekintett attól a kötelezettségtől, hogy benyújtja a Tarceva-val végzett vizsgálatok eredményeit a gyermekpopuláció minden alcsoportjában a nem kissejtes tüdőrák és a hasnyálmirigyrák indikációi tekintetében (lásd a 4.2 pontot a „gyermekgyógyászati alkalmazásra vonatkozó információkért”).
05,2 "Farmakokinetikai tulajdonságok
AbszorpcióAz erlotinib plazma csúcskoncentrációja körülbelül 4 órával az orális beadás után érhető el. Egy egészséges, egészséges önkéntesekkel végzett vizsgálat 59%-os becsült abszolút biohasznosulást mutatott. Az élelmiszer orális adag után növelheti az expozíciót.
terjesztés: Az erlotinib átlagos látszólagos eloszlási térfogata 232 l, és eloszlik az emberi daganatszövetben. Egy vizsgálatban, amelyet 4 betegen végeztek (3 nem kissejtes tüdőrákos (NSCLC) és 1 gégerákos), napi 150 mg Tarceva-val szájon át, a mintákat a tumor sebészeti kimetszésével nyerték a 9. napon. A kezelés során az erlotinib-koncentráció a tumoron belül átlagosan 1,185 ng / g szövetet mutatott, ami összességében az egyensúlyi állapotban megfigyelt csúcskoncentráció átlagos 63% -ának (tartomány: 5-161%) felel meg. Főbb metabolitok a tumorban, átlagosan 160 ng / g szövetkoncentrációban, ami összességében az egyensúlyi állapotban megfigyelt csúcs plazmakoncentráció 113%-ának (tartomány: 88-130%) felel meg, A plazmafehérjékhez való kötődés körülbelül 95%. Az erlotinib kötődik a szérumalbuminhoz és az alfa-1 savas glikoproteinhez (AAG).
BiotranszformációEmberben az erlotinibet a májban a máj citokrómjai metabolizálják, főként a CYP3A4 és kisebb mértékben a CYP1A2. az erlotinib aránya.
Három fő metabolikus utat azonosítottak: 1) az egyik vagy mindkét oldallánc O-demetilezése, majd karbonsavakká történő oxidáció; 2) az acetilénfrakció oxidálása, majd hidrolízis aril-karbonsavvá; és 3) a fenil-acetilén aromás hidroxilezése töredék. A fő erlotinib metabolitok, az OSI-420 és az OSI-413, amelyeket az egyik oldallánc O-demetilezésével állítottak elő, hasonló hatást mutattak az erlotinibhez, mint a nem klinikai elemzések. in vitro és tumor modellekben in vivo. Ezek a plazmában az erlotinib szintjének kevesebb, mint 10% -ánál vannak jelen, és hasonló farmakokinetikával rendelkeznek, mint az erlotinib.
Kiküszöbölés: az erlotinib főként a széklettel metabolizálódik (> 90%), míg a vesén keresztül történő elimináció az orálisan beadott mennyiségnek csak kis hányadát (kb. 9%-át) teszi ki. Az orálisan adagolt adag kevesebb, mint 2%-a ürül változatlan formában. Egy populáció farmakokinetikai elemzése A Tarceva monoterápiával kezelt 591 beteg közül az átlagos látszólagos clearance 4,47 l / óra, átlagos felezési ideje 36,2 óra, ezért az egyensúlyi plazmakoncentráció körülbelül 7 vagy 8 nap alatt várható.
Farmakokinetika speciális populációkban:
A populáció farmakokinetikai elemzése alapján nem észleltek klinikailag szignifikáns összefüggést a látszólagos clearance és a beteg kora, testtömege, neme vagy etnikai hovatartozása között. A beteggel kapcsolatos tényezők, amelyek összefüggést mutattak az erlotinib farmakokinetikájával, a teljes bilirubin, az A szérum összbilirubin- és AAG -koncentrációjának emelkedése az erlotinib -clearance csökkenésével járt. Ezen különbségek klinikai jelentősége nem világos. A dohányosoknál azonban megnőtt az erlotinib clearance aránya.
Ezt megerősítette egy farmakokinetikai vizsgálat egészséges, nem dohányzó és cigarettázó személyeken, akik egyetlen 150 mg-os orális erlotinb adagot kaptak. A Cmax geometriai átlaga nem dohányzókban 1056 ng / ml, dohányosoknál 689 ng / ml volt, a dohányzók és a nemdohányzók átlagos aránya 65,2% (95% CI: 44,3-95,9; p = 0,031). Az AUC0 -inf geometriai átlaga 18726 ng • h / ml volt nemdohányzóknál és 6718 ng • h / ml dohányosoknál, átlagos aránya 35,9% (95% CI: 23,7 - 54, 3; p
A kulcsfontosságú III. Fázisú NSCLC-vizsgálatban a dohányosok 0,65 μg / ml (n = 16) egyensúlyi állapotú erlotinib plazmakoncentrációt értek el, körülbelül kétszer alacsonyabbak, mint a korábbi dohányosok vagy soha nem dohányzó betegek (1,28 mcg / ml, n = 108). Ezt a hatást az erlotinib látszólagos plazma clearance -ének 24% -os növekedése kísérte. Az I. fázisú dózisnövelési vizsgálatban dohányzó NSCLC-s betegekben az egyensúlyi állapotú farmakokinetikai elemzések az erlotinib-expozíció dózisarányos növekedését mutatták, amikor a Tarceva adagját 150 mg-ról a maximális tolerálható 300-as dózisra emelték. Ebben a vizsgálatban az dohányosok 300 mg -os dózisánál a plazmakoncentráció 1,22 μg / ml volt (n = 17).
A farmakokinetikai vizsgálatok eredményei alapján a jelenlegi dohányosoknak azt kell tanácsolni, hogy hagyják abba a dohányzást a Tarceva szedése alatt, különben a plazmakoncentráció csökkenhet.
A populáció farmakokinetikai elemzése alapján úgy tűnik, hogy egy opioid jelenléte körülbelül 11%-kal növeli az expozíciót.
Egy második populációs farmakokinetikai elemzést végeztek, beleértve az erlotinib adatait 204 hasnyálmirigyrákos betegtől, akiket erlotinibdel és gemcitabinnel kezeltek. Ez az elemzés azt mutatta, hogy a hasnyálmirigyrákos betegeknél az erlotinib clearance -et befolyásoló kovariantok nagyon hasonlítottak a monoterápia korábbi farmakokinetikai elemzésében megfigyeltekhez. Új kovariáns hatásokat nem azonosítottak. A gemcitabin egyidejű alkalmazása nem befolyásolta az erlotinib plazma clearance-ét.
Gyermekpopuláció: Gyermekgyógyászati betegeken nem végeztek specifikus vizsgálatokat
Idős betegek: Idős betegeken nem végeztek specifikus vizsgálatokat.
Májelégtelenség: az erlotinib clearance -e túlnyomórészt máj. Szolid tumorokban és mérsékelt májkárosodásban szenvedő betegeknél (Child-Pugh pontszám 7-9) az erlotinib geometriai átlagos AUC0-t és Cmax értéke 27 000 ng • h / ml és 805 ng / ml, míg 29300 ng • h / ml és 1090 ng / ml megfelelő májfunkciójú betegeknél, beleértve az elsődleges májrákban vagy májmetasztázisban szenvedőket is. Bár a Cmax statisztikailag szignifikánsan alacsonyabb volt a közepesen súlyos májműködésű betegeknél, ez a különbség nem tekinthető klinikailag relevánsnak. a súlyos májműködési zavar hatása az erlotinib farmakokinetikájára. A populáció farmakokinetikai elemzésében a szérum összes bilirubin koncentrációjának növekedése az erlotinib clearance lassulásával jár.
Veseelégtelenség: Az erlotinib és metabolitjai vesén keresztül történő kiválasztása nem jelentős, mivel az egyszeri adag kevesebb mint 9% -a ürül a vizelettel. Klinikailag szignifikáns összefüggés az erlotinib -clearance és a kreatinin -clearance között, de nincsenek adatok a kreatinin -clearance -szel rendelkező betegekről
05.3 A preklinikai biztonságossági adatok
A legalább egy állatfajban vagy vizsgálatban megfigyelt hatások közül a szaruhártyán (sorvadás, fekélyek), a bőrön (tüsző degeneráció és gyulladás, bőrpír és alopecia), a petefészken (sorvadás), a májban (májelhalás), a vesében (vese papilláris nekrózis és tubuláris tágulat) és a gyomor -bél traktusban (késleltetett gyomorürülés és hasmenés). neutrofilek: az ALT, az AST és a bilirubin emelkedését hozták összefüggésbe, és ezek az adatok a klinikailag relevánsnál jóval alacsonyabb expozíciókra vonatkoztak.
A hatásmechanizmus alapján az erlotinib potenciálisan teratogén. Patkányokon és nyulakon végzett reprodukciós toxicitási vizsgálatokból származó adatok a maximális tolerált dózishoz közeli dózisokban és / vagy anyai toxikus dózisok reprodukciós toxicitást (embriótoxicitás patkányban) jeleztek., Embrionális felszívódást és magzati toxicitást nyulaknál) és a fejlődés (a kölykök növekedése és túlélése patkányokban), de nem mutattak teratogenitást vagy a termékenység károsodását. Ezeket az eredményeket klinikailag releváns expozíciók esetén figyelték meg.
Az erlotinib hagyományos genotoxicitási vizsgálatai sikertelenek. Patkányokon és egereken végzett 2 éves karcinogenitási vizsgálatok, amelyek során az erlotinib az emberi terápiás koncentrációt meghaladó koncentrációig (legfeljebb kétszeres, illetve 10-szeres, a Cmax és / vagy AUC alapján) negatív volt.
Patkányokban enyhe fototoxikus bőrreakciót figyeltek meg az UV besugárzás után.
06.0 GYÓGYSZERÉSZETI INFORMÁCIÓK
06.1 Segédanyagok
A tabletta magja:
Laktóz -monohidrát
Mikrokristályos cellulóz (E460)
A típusú nátrium -keményítő -glikolát
Nátrium -lauril -szulfát
Magnézium -sztearát (E470 b)
Tabletta bevonat:
Hidroxi -propil -cellulóz (E463)
Titán -dioxid (E171)
Makrogol
Hipromellóz (E464)
06.2 Inkompatibilitás
Nem releváns.
06.3 Érvényességi idő
4 év.
06.4 Különleges tárolási előírások
Ez a gyógyszer nem igényel különleges tárolási körülményeket.
06.5 A közvetlen csomagolás jellege és a csomag tartalma
PVC buborékcsomagolás alumínium fóliával, 30 tablettát tartalmaz.
06.6 Használati utasítás
Nincsenek különleges utasítások a megsemmisítésre.
A fel nem használt gyógyszert és a gyógyszerből származó hulladékot a helyi előírásoknak megfelelően kell megsemmisíteni.
07.0 FORGALOMBA HOZATALI ENGEDÉLY
Roche Registration Limited
6 Sólyom út
Shire Park
Welwyn kertváros
AL7 1TW
Egyesült Királyság
08.0 A FORGALOMBA HOZATALI ENGEDÉLY SZÁMA
EU/1/05/311/003
036871034
09.0 Az első forgalomba hozatali engedély kiadásának időpontja
Az első engedélyezés időpontja: 2005. szeptember 19
Az utolsó megújítás dátuma: 2010. szeptember 19
10.0 A SZÖVEG FELÜLVIZSGÁLÁSÁNAK DÁTUMA
2016. január
















-cos-cause-sintomi-e-cura.jpg)