Általánosság
Az Osteogenesis imperfecta veleszületett genetikai betegség, amely nem kötődik a nemhez, felelős a csontok bizonyos törékenységéért és a törések hajlamának.
Az osteogenesis imperfecta tünetei számosak; általában a következőkből állnak: csontgyengülés, nagy csonttörési hajlam, kék, szürke vagy lila szemhéjak, csontdeformitások vagy egyéb csontváz elváltozások, háromszög alakú arc, fogászati törékenység stb. .
Általánosságban elmondható, hogy az osteogenesis imperfecta helyes diagnosztizálásához elengedhetetlenek a következők: fizikális vizsgálat, kórtörténet, orvosi képalkotó vizsgálatok, I. típusú kollagénvizsgálati teszt és genetikai vizsgálat.
Sajnos jelenleg az osteogenesis imperfecta betegek számára csak a tüneti kezelések állnak rendelkezésre. A szóban forgó betegség valójában gyógyíthatatlan.
Mi az osteogenesis imperfecta?
Az osteogenesis imperfecta genetikai betegség, amely az érintett személy csontjait gyengébbé és hajlamosabbá teszi a törésekre.
A valóságban az osteogenesis imperfecta kifejezéssel az orvosok a genetikai betegségek heterogén csoportjára utalnak, amelyet a csontok bizonyos fokú törékenysége jellemez. Ezért az osteogenesis imperfecta számos formája (vagy típusa) létezik, amelyek közül néhány sokkal súlyosabb, mint mások.
Ez egy veleszületett betegség
Azoknál az embereknél, akiket ez érint, az osteogenesis imperfecta a születéstől fogva jelen lévő betegség, ezért minden értelemben veleszületett betegségként határozható meg.
SEX KAPCSOLATOS?
Az Osteogenesis imperfecta nem nemi eredetű genetikai betegség, például hemofília vagy Klinefelter-szindróma.
EPIDEMILÓGIA
Egyes statisztikai kutatások szerint az osteogenesis imperfecta előfordulási gyakorisága 15 000-20 000 születés esetén egyenlő lenne. Ez azt jelenti, hogy minden 15 000–20 000 újszülöttnek van osteogenesis imperfecta.
Más statisztikai vizsgálatok azt is kimutatták, hogy az osteogenesis imperfecta egyformán érinti a férfiakat és a nőstényeket, és nem részesíti előnyben egy adott populációt vagy etnikai csoportot.
Az élettartam rendkívül változó paraméter, amely az osteogenesis imperfecta formájától függ.
Okoz
Az Osteogenesis imperfecta szinte mindig az I. típusú kollagén termelésének minőségi és mennyiségi megváltozásából ered.
Az I. típusú kollagén elengedhetetlen a csontok erősítéséhez és az egészséges kötőszövetek fenntartásához, beleértve a porcokat, inakat, bőrt, szemhéjat stb.
Ezért az I. típusú kollagén termelésének megváltozása befolyásolja a csontok erejét és az emberi testben lévő kötőszövetek jó egészségét.
MI VÁLTOZTATJA A KOLLAGÉNTermelést?
A genetikai betegség olyan állapot, amely a sejt DNS -t alkotó egy vagy több gén mutációja miatt keletkezik.
Az osteogenesis imperfecta esetében az utóbbinak okai szinte mindig a COL1A1 (a 17. kromoszómán található) és a COL1A2 (a 7. kromoszómán található) gén egyikének vagy mindkettőjének mutációjában kereshetők.
Normál körülmények között a COL1A1 és a COL1A2 szabályozza az I. típusú kollagén normál termelését; töltésükben lévő mutációk jelenlétében megbuknak szabályozó funkciójukban.
Fontos: milyen más gének, ha mutálódnak, osteogenesis imperfectát okoznak?
A COL1A1 és COL1A2 mutációk mellett az IFITM5, SERPINF1, CRTAP és LEPRE1 gének mutációi az osteogenesis imperfecta lehetséges okai.
A fent említett gének a COL1A1 -től és a COL1A2 -től eltérő funkciókat fednek le - ezért nem szabályozzák az I. típusú kollagén termelését -, de még mindig "befolyásolják az emberi csontváz csontjainak erejét és ellenállását.
MILYEN GENETIKUS BETEGSÉG?
Az osteogenesis imperfecta autoszomális genetikai betegség.
Az genetikai betegséghez társuló autoszomális kifejezés azt jelzi, hogy a szóban forgó állapot az autoszomális és nem nemi kromoszómákon alapuló genetikai mutációknak köszönhető.
Felhívjuk az olvasók figyelmét arra, hogy az ember 23 pár kromoszómából álló kromoszóma -készlettel rendelkezik, amelyekben 22 pár autoszomális típusú, és csak egy pár szexuális típusú. A szexuális típusú kromoszómapár befolyásolja a nemet egyéni.
A COL1A1, a COL1A2 és az IFITM5 mutációit követő osteogenesis imperfecta rendelkezik az autoszomális domináns betegség összes jellemzőjével, amikor a SERPINF1, CRTAP és LEPRE1 gének mutációinak köszönhető, akkor az autoszomális recesszív betegség jellemzőivel rendelkezik.
TÍPUSOK
Jelenleg az orvosok úgy vélik, hogy 8 típusú (vagy formájú) osteogenesis imperfecta létezik. A különböző típusok megkülönböztetése érdekében úgy döntöttek, hogy a római számozást használják, pontosabban az első nyolc római számot.
Az alábbi táblázat az osteogenesis imperfecta 8 formáját, az őket okozó mutációkat és egyéb jellemzőket mutatja be.
Fickó
Mutált gén
A genetikai betegség típusa
AZ
COL1A1
Autoszomális domináns
II
COL1A1 és COL1A2
Autoszomális domináns
III
COL1A1 és COL1A2
Autoszomális domináns
IV
COL1A1 és COL1A2
Autoszomális domináns
V.
IFITM5
Autoszomális domináns
TE
SERPINF1
Autoszomális recesszív
VII
CRTAP
Autoszomális recesszív
VIII
HARE 1
Autoszomális recesszív
* Megjegyzés: nyilvánvaló, hogy a COL1A1 és a COL1A2 mutációi, amelyek az osteogenesis imperfecta első négy formáját okozzák, genetikai elváltozások, kissé eltérő jellemzőkkel. Különben nincs értelme megkülönböztetni egyiket a másiktól.
Tünetek, jelek és szövődmények
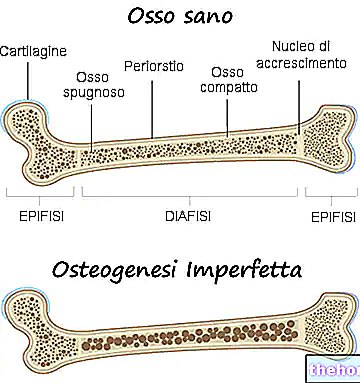
Az osteogenesis imperfecta minden típusa felelős a csontok gyengüléséért, így a betegség által érintett személy különösen hajlamos a törésekre. A csontok gyengülésének mértéke az alaktól függően változik; ezek közül néhánynak ez a gyengülés nagyobb, mint másoknak.
Ezt követően meg kell jegyezni, hogy az osteogenesis imperfecta minden formájának saját tüneti képe van, amely egyesek számára felidézheti más formák tüneti képét.
LEHETSÉGES TÜNETEK ÉS JELEK
Az osteogenesis imperfecta lehetséges tünetei és jelei a következők:
- Csonthibák jelenléte;
- Rövid és kicsi test (törzsnek szánva);
- Ízületi problémák (pl. Laza ízületek);
- Izomgyengeség;
- Kék, lila vagy szürke szemhéj;
- Háromszög alakú arc;
- Hordó láda;
- A gerincoszlop morfológiai anomáliái;
- Fogászati törékenység;
- A hallás csökkenése vagy teljes elvesztése;
- Légzési gondok
- Az 1 -es típusú kollagén hiányával vagy hiányával kapcsolatos problémák.
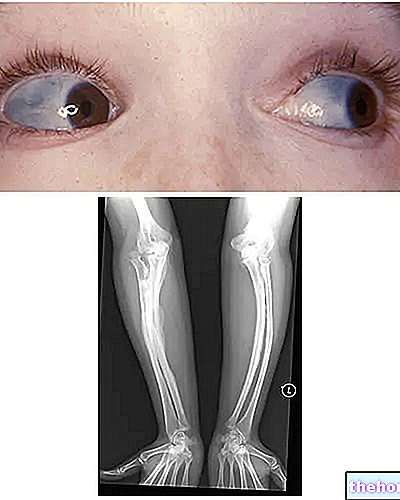
Osteogenesis Imperfecta: vegye figyelembe a sclerae kék színét és a betegséget jellemző csontdeformációkat. A wikipedia.org oldalról
MILYEN A HIBA TÖKÉLETES OSTEOGENÉZIS LEGKOMOLYABB FORMÁJA?
Az orvosok az osteogenesis imperfecta különféle típusainak tüneti súlyosságát 3 fokos skálán osztályozzák, amelyek a következők: enyhe, közepes fokú és súlyos fokú.
Csak egy forma tartozik az "enyhe fok" kategóriába: "I. típusú osteogenesis imperfecta"; 4 osteogenesis imperfecta forma a "mérsékelt fokú" kategóriába tartozik: IV, V és VI; végül a "súlyos fokú" kategória 3 formák: II, III, VII és VIII.
I. TÍPUS: JELLEMZŐK
A leggyakoribb és legkevésbé súlyos forma, az I. típusú osteogenesis imperfecta a következő jellemzőkkel rendelkezik:
- Különösen a pubertás előtt okoz töréseket;
- Ez "szinte semmilyen hatással nincs a magasságra, ezért a betegek általában normális magasságúak";
- Izomproblémákat és izomgyengeséget okoz
- Felelős a kék, lila vagy szürke skleráért;
- Ez az arc és a gerinc háromszög alakú anomáliáinak oka;
- Szinte soha nem okoz csontdeformációt. Ha provokálja őket, akkor minimálisak;
- Fogászati törékenységet és / vagy halláskárosodást okozhat (ez utóbbi általában felnőttkorban fordul elő);
- Az I. típusú kollagén jelenlétével van összefüggésben, amely minőségben normális, de mennyiségben abnormális (a normálisnál gyengébb).
II. TÍPUS: JELLEMZŐK
A II. Típusú osteogenesis imperfecta -t a következők jellemzik:
- Halál oka születéskor vagy röviddel azután. A légzési problémák szinte mindig halált okoznak;
- Jelentős csonttörékenység és súlyos csontdeformitások jelenléte;
- Rövid termetű és fejletlen tüdő
- Kék, lila vagy szürke színű sclera;
- Az I. típusú kollagén mennyiségi és minőségi anomáliáinak jelenléte.
III. TÍPUS: JELLEMZŐK
A III. Típusú osteogenesis imperfecta a következő jellemzőkkel rendelkezik:
- Bár nagyon súlyos, nem gyakran okoz halált az újszülött korban;
- A "nagy csonttörékenységgel társul;
- Felelős az alacsony termetért, az ízületi problémákért, az izomgyengeségért (különösen a lábakban és a karokban), a hordó mellkasáért, a háromszög alakú arcért és a gerinc kóros görbületéért;
- Ennek oka a kék, lila vagy szürke sclera;
- Légzési problémákat, fogászati törékenységet és halláskárosodást okozhat;
- Gyakran felelős a csont deformitásáért;
- Az I. típusú kollagén minőségi és mennyiségi rendellenességeihez kapcsolódik.
IV. TÍPUS: JELLEMZŐK
A IV. Típusú osteogenezist a következők jellemzik:
- A csontok törékenységének egy foka a II. És a III. Forma és az I. forma között;
- Az átlagosnál alacsonyabb termetű;
- Kék, lila vagy szürke színű sclera;
- Enyhe / közepes súlyosságú csontdeformitások, a gerinc és a hordó mellkasának enyhe rendellenességei;
- Háromszög alakú arc;
- Fogászati törékenység és halláskárosodás lehetséges jelenléte;
- I. típusú kollagén rendellenességek jelenléte.
V. TÍPUS: JELLEMZŐK
Az V. típusú osteogenesis imperfecta bizonyos szempontból hasonlít a IV. Típusú osteogenesis imperfecta -ra. Van azonban néhány sajátossága, amelyek a következők:
- Normál színű sclera;
- Fogászati törékenység hiánya;
- Rendellenes csontos bőrkeményedések kialakulása a törött csontok gyógyulási folyamata során;
- A sugár és az ulna között elhelyezkedő interosseous membrán meszesedése. Ez rontja az alkar mobilitását.
VI. TÍPUS: JELLEMZŐK
A VI típusú osteogenesis imperfecta is hasonló a IV formához. Az utóbbitól való megkülönböztetésre néhány sajátosság, köztük az alkalikus foszfatáz magas vérszintje, és egyes csontoknál a halak tüskéihez hasonló lamellák (csontos) jelenléte.
VII. TÍPUS: JELLEMZŐK
Tünetileg a VII -es típusú osteogenesis imperfecta bizonyos körülmények között hasonlíthat a IV -es típushoz, más esetekben a II.
Ennek a súlyos kóros formának a sajátosságai a következők:
- Alacsony termetű;
- Rendkívül rövid humerus (karcsont) és combcsont (combcsont) jelenléte;
- A csípő deformitásának gyakori jelenléte, coxa vara néven.
VIII. TÍPUS: JELLEMZŐK
A VIII. Típusú osteogenesis imperfecta nagyon emlékeztet a II. És III. Formára.
Különös jellemzői közül kiemelkednek a következők: súlyos növekedési hiány, súlyos csontváz-hipomineralizáció és a prolil-3-hidroxiláz enzim hiánya (vagy szűkös jelenléte).
Diagnózis
Általában az a diagnosztikai folyamat, amelyet az osteogenesis imperfecta feltételezett formájával rendelkező betegeknek vetnek alá, gondos fizikális vizsgálattal és gondos kórtörténettel kezdődik; majd folytatódik, "a beteg családtörténetének elemzésével és számos diagnosztikai képalkotó teszttel (röntgen, CT-vizsgálat stb.); végül az I. típusú kollagén mennyiségi és minőségi értékelésével, valamint genetikai teszt.
Manapság lehetőség van az osteogenesis imperfecta diagnosztizálására még a prenatális fázisban is, ha egy terhes nőt ultrahangnak vetnek alá.
A CÉLKITŰZÉS ÉS A TÖRTÉNET FONTOSSÁGA
Az osteogenesis imperfecta orvos szakértője nagyon gyakran képes csak a fizikális vizsgálat és az anamnézis segítségével diagnosztizálni a fent említett betegséget. Ez azt jelenti, hogy ezeknek a diagnosztikai vizsgálatoknak nem elhanyagolható jelentőségük van.
AZ I. TÍPUSÚ KOLLAGÉNTermelés értékelése
Általános szabály, hogy az I. típusú kollagén minőségi és mennyiségi értékelése nagyon megbízható teszt, mivel, mint említettük, az osteogenesis imperfecta esetek többségét az 1 -es típusú kollagén termelését szabályozó gének mutációi jellemzik.
Az egyének sejtszinten jelen lévő I. típusú kollagén mennyiségének és minőségének felméréséhez az orvosok bőrbiopsziára vagy egy adott vérvizsgálatra támaszkodhatnak.
Mindkét értékelő teszt meglehetősen összetett, és a páciensnek (vagy szüleinek) több hetet kell várnia az eredmények megismerésére.
GENETIKAI VIZSGÁLAT
Egy genetikai teszt segítségével, amely a vizsgált egyed teljes DNS -jét vizsgálja, az orvosok véglegesen meghatározhatják a jelenlévő genetikai mutáció jellemzőit.
Általában minden sejt DNS -en genetikai vizsgálatot kell végrehajtani, ha az I. típusú kollagén jellemzőinek értékelése nem hozta meg a kívánt eredményeket, vagy ha nem a COL1A1 vagy a COL1A2 mutációja okozza az "osteogenesis imperfecta -t".
PRENATÁLIS DIAGNÓZIS
A prenatális ultrahang nagyon hasznos a II és III típusú osteogenesis imperfecta azonosítására.
Terápia
Jelenleg nincs specifikus gyógymód az osteogenesis imperfecta -ra, más szóval, az osteogenesis imperfecta -ban szenvedőknek az a rendeltetésük, hogy halálukig együtt élnek a fent említett állapottal, ami gyakran maga a betegség következményeinek köszönhető.
A specifikus terápia hiánya nem zárja ki más kezelési formák létezését. Valójában az osteogenesis imperfecta -ban szenvedő beteg terápiás lehetőségei között különféle tüneti terápiák szerepelnek; tüneti terápiák alatt olyan kezeléseket értünk, amelyek képesek a tünetek enyhítésére, a betegség lefolyásának lelassítására és a legsúlyosabb következmények megelőzésére (vagy legalább elhalasztására).
LEHETSÉGES TÜNETI KEZELÉSEK
Az osteogenesis imperfecta lehetséges tüneti kezeléseinek listájában a következők emelkednek ki:
- A körmök sebészeti behelyezése a leghosszabb csontok belsejébe (N.B .: a leghajlamosabbak a törésekre), amelyek nagyobb ellenállást biztosítanak a törésekkel és deformitásokkal szemben. Ezt a műveletet ún rúddobálás intramedulláris;
- Törések és / vagy csontdeformitások konzervatív vagy sebészeti kezelése;
- Fogápolás, a fogak egészségének megőrzése érdekében;
- Fájdalomcsillapító terápiák, nagyon fájdalmas többszörös törések esetén;
- Fizioterápia, izomhosszabbításhoz és erősítéshez Az elasztikus és tónusos izomberendezés lehetővé teszi, hogy megelőzze az eséseket, amelyek különböző csonttörésekhez vezethetnek;
- Segédeszközök használata a mozgáshoz, beleértve a kerekesszékeket, merevítőket, mankókat stb.
A MOZGÁS ELŐNYEI
Az osteogenesis imperfecta betegeknél az orvosok a fizikai gyakorlatok és általában a mozgás folyamatos gyakorlását javasolják, mivel mindkettő hozzájárul a csont- és izomrendszer megerősítéséhez.
Az ajánlott sportágak közé tartozik: úszás, mivel ez „csontvázra gyakorolt alacsony fizikai aktivitás”, és gyaloglás.
ELŐNYÖK AZ EGÉSZSÉGES életmódból
Az egészséges életmód, a dohányzás elkerülése, a túlzott alkoholfogyasztás, a túlzott és rossz étkezés stb. Több mint diszkrét egészségügyi előnyökkel jár az osteogenesis imperfecta betegek számára, mivel lassítja a betegség előrehaladását és csökkenti a csontok törékenységét.
TÜNETI KEZELÉSEK A KÍSÉRLETI FÁZISBAN
Jelenleg az orvosok és kutatók értékelik egyes tüneti kezelések hatékonyságát, beleértve a növekedési hormon kezelést és a biszfoszfonát alapú intravénás és orális terápiát.
Jelenleg a fent említett vizsgálati kezelések által nyújtott eredmények az egész orvosi közösség számára jót ígérnek.
Prognózis
Az osteogenesis imperfecta negatív prognózisú betegség, mivel gyógyíthatatlan, drasztikusan rontja az életminőséget, és bizonyos esetekben az érintett korai halálát okozza.
Meg kell azonban jegyezni, hogy a modern tüneti kezeléseknek is köszönhetően sok egyén, akiknek enyhe formájú osteogenesis imperfecta van, kellemes és kielégítő életet élhetnek.
Megelőzés
Sajnos jelenleg nincs megelőző intézkedés az osteogenesis imperfecta ellen.